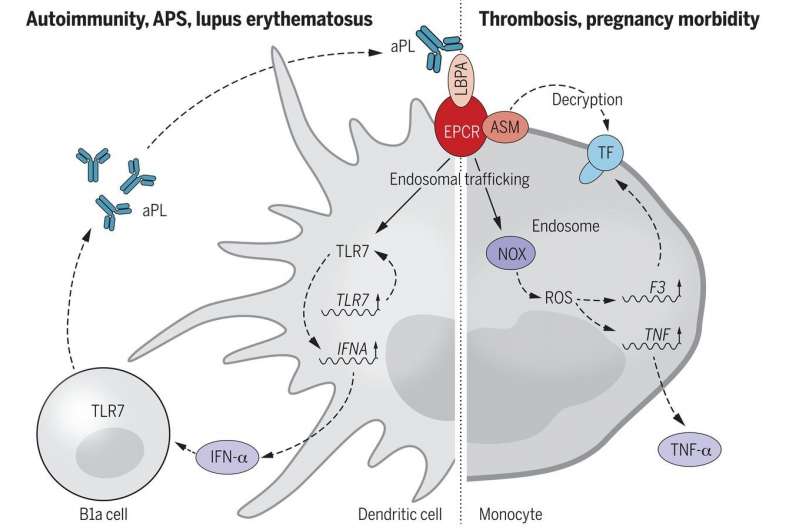
(Sindrome da antifosfolipidi-Immagine:il riconoscimento di EPCR-LBPA da parte di aPL promuove l’autoimmunità. Credito: Science (2021). DOI: 10.1126 / science.abc09569).
Un team di ricercatori affiliati a più istituzioni in Germania e negli Stati Uniti ha trovato un bersaglio sulla superficie cellulare per gli autoanticorpi che prendono di mira i fosfolipidi (aPL) che contribuiscono alla sindrome da antifosfolipidi (APS). Nell’articolo pubblicato sulla rivista Science, il gruppo descrive come ha utilizzato modelli murini di APS con perdita fetale indotta di aPL e trombosi per studiare la connessione tra i fattori che contribuiscono all’APS.
L’APS è una malattia autoimmune che espone i pazienti a un rischio maggiore di una varietà di disturbi che vanno dalla trombosi alle complicanze ostetriche. Tende a verificarsi in pazienti che soffrono di condizioni di autoimmunità come il lupus. La ricerca precedente ha portato alla scoperta di marcatori che possono essere utilizzati per identificare la sindrome da antifosfolipidi nei pazienti, ma non è ancora noto il motivo per cui si verifica la condizione. Uno di questi marker è un aumento della presenza di autoanticorpi che prendono di mira i fosfolipidi. Sfortunatamente, c’è anche un problema quando si identifica la ragione alla base di un aumento di tali livelli: anche altre circostanze possono farli aumentare, come le malattie infettive. In questo nuovo sforzo, i ricercatori si sono concentrati solo sui casi in cui i livelli elevati sono stati associati alla sindrome da antifosfolipidi.
Vedi anche:Sindrome antifosfolipidica, come i comuni batteri intestinali innescano la malattia
Studiando il recettore della proteina C endoteliale (EPCR) e il modo in cui funziona con l’acido lisobisfosfatidico (LBPA), i ricercatori sono stati in grado di identificare i bersagli della superficie cellulare per aPL e vedere come era mediato internamente. Più specificamente, hanno scoperto che EPCR fungeva da recettore della superficie cellulare per APL. Hanno anche scoperto che EPCR mediava l’internalizzazione di aPL in determinate circostanze. E hanno scoperto che il legame di aPL con EPCR-LBPA ha portato all’attivazione della coagulazione e alla produzione di interferone-α nelle cellule dendritiche. E questo ha portato alla creazione di più cellule B1a, che sono i produttori di aPL. I ricercatori hanno scoperto che il blocco del legame EPCR-LBPA nei modelli murini ha impedito lo svolgersi della catena di eventi che ha portato a una riduzione dei livelli di aPL.
Spiegano gli autori:
“Gli anticorpi antifosfolipidi (aPL) compaiono transitoriamente nelle malattie infettive ma possono persistere nella sindrome da antifosfolipidi primari (APS) o in associazione con altre malattie autoimmuni, incluso il lupus eritematoso. Le manifestazioni cliniche della APS vanno dalla trombosi venosa e ictus a gravi danni agli organi microangiopatici e complicanze della gravidanza. L’attivazione della coagulazione e delle cascate del complemento è cruciale per gli effetti patogeni degli aPL e per la loro segnalazione cellulare proinfiammatoria nelle cellule trofoblastiche immunitarie innate, endoteliali vascolari ed embrionali. Tuttavia, la diversa reattività di aPL con lipidi caricati negativamente e con proteine del sangue ha ostacolato l’identificazione dei meccanismi centrali alla base della segnalazione patogena di aPL. Gli aPL reattivi ai lipidi sensibilizzano le cellule immunitarie innate agli agonisti del recettore 7 (TLR7) e regolano il fattore tissutale iniziatore della coagulazione (TF). La segnalazione TF-dipendente dal recettore 2 attivato dalla proteasi (PAR2) coinvolge il recettore della proteina C endoteliale (EPCR, codificato da PROCR ) e questo complesso di segnalazione partecipa specificamente alle risposte dell’interferone (IFN) a valle di TLR4. Poiché gli aPL sono potenti induttori della produzione di IFN-α da parte delle cellule dendritiche, abbiamo testato l’ipotesi che EPCR contribuisca alle patologie APS e allo sviluppo dell’autoimmunità aPL”.
Fonte:Science