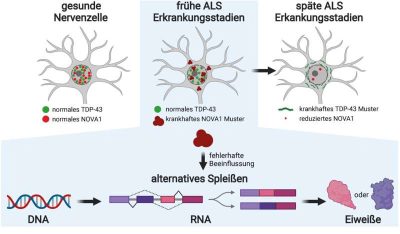
(SLA-Immagine: fasi di sviluppo in una cellula nervosa affetta da SLA. Credito: Prof. Dr. Beate Wimmer).
Attualmente non esiste una cura per la sclerosi laterale amiotrofica (SLA). Le cose potrebbero presto cambiare, però. I ricercatori della FAU e dell’Università della California di San Diego (UCSD) hanno identificato una proteina che mostra già caratteristiche patologiche in una fase iniziale della malattia neurologica SLA.
Il team ha pubblicato la sua scoperta, che potrebbe portare a un nuovo approccio per il trattamento della malattia, sulla rivista Acta Neuropathologica.
Nell’estate 2014, la sclerosi laterale amiotrofica o SLA, ha ricevuto molta attenzione attraverso una campagna sui social media. In Germania, circa 6.000-8.000 persone convivono con la SLA e ogni anno vengono diagnosticati circa 2.000 nuovi casi della malattia, che si rivela fatale nel giro di pochi anni. “La SLA è una malattia dei motoneuroni, ciò significa che danneggia le cellule nervose che controllano i nostri muscoli“, spiega il Prof. Dr. Beate Winner. “Durante la prima fase, i muscoli si indeboliscono, prima di deperire e, infine, lasciare i pazienti incapaci di deglutire o respirare autonomamente“.
Riportare indietro l’orologio biologico riprogrammando le cellule in cellule staminali
Beate Winner è Professore di modelli di cellule staminali per malattie neurali rare presso la FAU, capo del Dipartimento di biologia delle cellule staminali e relatore per il Center for Rate Diseases presso l’Universitätsklinikum Erlangen. Il suo laboratorio indaga su cosa scatena le malattie neurodegenerative del sistema nervoso come la SLA nella speranza di scoprire nuove opzioni di trattamento.
“Sappiamo da circa 15 anni che durante la fase terminale della SLA, la proteina TDP-43 che si trova nei neuroni diventa insolubile e inizia a formare grumi”, spiega Winner. “Perde le sue normali funzioni e adotta proprietà tossiche“. Anche se questi cambiamenti patologici non sono ancora evidenti nei pazienti, il destino delle cellule nervose è già segnato. “Volevamo sapere se potevamo trovare le cause della SLA in una fase iniziale di sviluppo prima che TDP-43 cambiasse”.
Vedi anche:SLA: farmaco sperimentale più efficace degli esistenti
Winner ha iniziato la sua ricerca insieme al Prof. Dr. Jürgen Winkler e al PD Dr. Martin Regensburger del Dipartimento di Neurologia Molecolare dell’Universitätsklinikum Erlangen. I ricercatori hanno utilizzato una tecnica innovativa. Hanno estratto un piccolo campione di pelle dalla parte superiore del braccio di pazienti affetti da SLA e persone sane in un gruppo di controllo e lo hanno riprogrammato in quelle che sono note come cellule staminali pluripotenti indotte, cellule che sono equivalenti a una fase molto precoce dello sviluppo umano e che in teoria possono svilupparsi in qualsiasi cellula del corpo umano. Queste cellule staminali sono state poi trasformate in cellule nervose. “Fondamentalmente, abbiamo riportato indietro l’orologio e generato neuroni che imitavano la fase di sviluppo di un feto“, spiega Winner. Il fatto che le cellule di persone adulte possano essere riprogrammate in cellule staminali pluripotenti è stato scoperto da Shin’ya Yamanaka, che ha ricevuto il Premio Nobel per la Medicina in riconoscimento del suo lavoro.
La proteina NOVA1 mostra caratteristiche patologiche sin dalle prime fasi
I ricercatori di Erlangen hanno cercato proteine insolubili nei campioni cellulari utilizzando la spettrometria di massa, una procedura ad alto rendimento. Hanno avuto successo. Nelle cellule nervose dei pazienti con SLA hanno scoperto una proteina legante l’RNA chiamata NOVA1. “Nei neuroni, la proteina ha mostrato cambiamenti tra cui un grado di insolvenza notevolmente aumentato, ma non ancora le caratteristiche patologiche tipiche del TDP-43″, spiega il Dottor Florian Krach, membro del team FAU e autore principale dello studio. “Le cellule nel gruppo di controllo non hanno visualizzato queste modifiche”.
Forte di questi risultati, Krach si è trasferito nel laboratorio del rinomato biologo e specialista in bioinformatica dell’RNA Prof. Gene Yeo presso l’Università della California a San Diego (USA), finanziato dal Bavaria California Technology Center (BaCaTeC). Grazie a esperimenti specializzati e analisi assistite da computer è stato in grado di indagare a cosa si lega NOVA1 nelle molecole di RNA e quale influenza ha sullo splicing alternativo nei neuroni umani. “Lo splicing alternativo è un meccanismo estremamente complesso e ingegnoso che gli esseri umani usano per moltiplicare il loro repertorio di proteine”, spiega Krach. “Sezioni di una molecola messaggero di RNA vengono tagliate o aggiunte, ostacolando, estendendo o modificando del tutto la funzione delle proteine“.
I ricercatori sperano che i loro risultati contribuiranno a rendere possibile una diagnosi precoce e apriranno la porta a nuovi trattamenti.
È noto da tempo che il processo di splicing alternativo non è regolamentato nei pazienti con SLA. È anche noto che TDP-43 influenza questo processo. Il team di ricercatori di Erlangen sospettava, tuttavia, che altre proteine leganti l’RNA fossero responsabili dei processi patologici nelle fasi iniziali della malattia prima che TDP-43 cambiasse. Questo sospetto è stato ora confermato con la scoperta del funzionamento alterato di NOVA1.
“Abbiamo fatto una scoperta pionieristica, ma è solo un primo passo verso la possibilità di essere in grado di rilevare la SLA nelle fasi iniziali”, afferma Beate Winner. “Studi di follow-up con coorti più ampie potrebbero approfondire la nostra comprensione dell’importanza delle proteine leganti l’RNA”.
Spiegano gli autori:
“La sclerosi laterale amiotrofica (SLA) è una malattia mortale caratterizzata da splicing alternativo (AS) aberrante. La perdita nucleare e l’accumulo citoplasmatico del fattore di splicing TDP-43 nei motoneuroni (MN) sono segni distintivi della SLA negli stadi avanzati della malattia. Tuttavia, non è noto se l’AS alterato sia presente prima che si verifichi la patologia TDP-43. Qui, indaghiamo l’AS alterato e le sue origini nelle prime fasi della SLA utilizzando motoneuroni (MN) derivati da cellule staminali pluripotenti indotti da pazienti con SLA sporadica e familiare. Troviamo alti livelli delle proteine leganti l’RNA NOVA1, NOVA2 e RBFOX2 nelle frazioni proteiche insolubili e osserviamo che gli eventi AS nei motoneuroni associati alla SLA sono arricchiti per i siti di legame di queste proteine. Il nostro studio indica una funzione interrotta precocemente di NOVA1 che guida i cambiamenti AS in modo complesso, compresi gli eventi causati da una perdita consistente della funzione NOVA1 che mostra un aumento dei livelli di proteine citoplasmatiche nei motoneuroni in fase iniziale senza patologia TDP-43 nel tessuto SLA post mortem. Quando il livello di proteina nucleare TDP-43 si esaurisce, NOVA1 si riduce…… Questo studio evidenzia che ulteriori perturbazioni RBP-RNA nella SLA si verificano parallelamente a TDP-43“.
I ricercatori sperano che il loro lavoro contribuisca allo sviluppo di nuovi concetti terapeutici prima che i neuroni attraversino il punto di non ritorno.
Fonte: Acta Neuropathologica