La scoperta riguarda difetti in una proteina chiamata FUS che svolge un ruolo importante nel traghettare materiali essenziali per la costruzione di proteine nelle cellule del cervello e del midollo spinale.
I ricercatori dell’Università di Toronto in Canada e Università di Cambridge nel Regno Unito, riferiscono le loro intuizioni sulla rivista Neuron.
Essi suggeriscono che lo studio potrebbe essere un importante passo avanti nella ricerca di un trattamento efficace per la sclerosi laterale amiotrofica (SLA) e demenza frontotemporale, una forma meno comune di demenza che danneggia i lobi frontali del cervello.
Autore senior dello studio è il e Professore Peter George-Hyslop St., uno scienziato medico, neurologo e genetista molecolare che guida la ricerca sulle malattie neurodegenerative in entrambe le università.
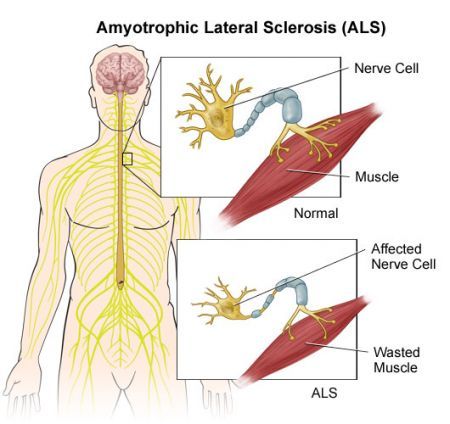
La SLA distrugge i motoneuroni, le cellule cerebrali che controllano il movimento dei muscoli.
Le persone con SLA gradualmente si indeboliscono e perdono la loro capacità di parlare, deglutire e respirare. .
In molti casi, sembra che la SLA è innescata da un accumulo di proteine tossiche che uccidono i neuroni nel cervello e nel midollo spinale.
Negli ultimi anni, i ricercatori hanno anche collegato la SLA a mutazioni nei geni che codificano per RNA-binding proteine - molecole che si attaccano all’ RNA (acido ribonucleico, che trasporta le informazioni genetiche copiate dal DNA) per controllare molti aspetti della regolazione genica: commutazione geni on/off o aumento e diminuzione della loro espressione. RNA è anche importante per la sintesi proteica nelle cellule.
Una proteina-RNA legame colpita da tali mutazioni – chiamata FUS – comunemente si accumula nel cervello e nel midollo spinale di pazienti con la SLA, ma non è mai stata considerata una delle cause della malattia, perché sembra diversa dai ciuffi chiaramente tossici delle proteine tau, amiloide e alfa sinucleina che si formano sempre in malattie come l’Alzheimer, il Parkinson e alcune forme di demenza.
Negli individui sani, FUS normalmente svolge un ruolo importante nel trasporto di prodotti chimici essenziali, segnalazione nel cervello e nel midollo spinale. Condivide una caratteristica unica con altri RNA-binding proteins.
La caratteristica distintiva è la capacità di trasformarsi da liquido in gel e di nuovoina liquido. Sembra che questa proprietà aiuta FUS ad accumulare materiali che le cellule nervose richiedono per produrre nuove proteine e depositarle in confezioni ordinate lungo il perimetro delle cellule.
Quando in transito, FUS assume la forma di un gel e quando deve spargere il suo carico, si trasforma in un liquido.
Questa caratteristica unica sembra essere importante per parti specifiche di cellule per ottenere i materiali precisi e necessari rapidamente ed efficientemente.
La squadra ha trovato che una mutazione nel gene che codifica per FUS riguarda la sua capacità di trasformarsi da un gel in un liquido. FUS mutato FUS causa forme molto spesse di “idrogeli fibrillari irreversibili” che non si trasformano di nuovo in liquidi. Il Prof. George-Hyslop St. spiega l’effetto che questo ha:
“Questo uccide il nervo impedendogli di produrre nuove proteine nelle parti della cellula che ne ha un disperato bisogno. Le mutazioni forzano il processo di gelificazione”.
I ricercatori ritengono che una soluzione per prevenire l’ over-ispessimento o invertire la solidificazione del gel potrebbe essere un passo avanti per un trattamento efficace per la SLA e demenza frontotemporale, malattie in cui le mutazioni in FUS portano a questo effetto.
La scoperta può anche fornire una via da seguire per altri tipi di SLA dove altri RNA-binding proteins gelificanti solidificano.
Lo studio segue una scoperta precedente, riportata nel mese di gennaio 2015 sulla rivista Cerebral Cortex, in cui i ricercatori spiegano di aver trovato un meccanismo cellulare che svolge un ruolo chiave nella SLA.
Fonte:
- Tetsuro Murakami et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron, October 2015 DOI:10.1016/j.neuron.2015.10.030