La sindrome di Angelman è una rara malattia genetica causata da mutazioni nel gene UBE3A ereditato dalla madre e caratterizzata da scarso controllo muscolare, linguaggio limitato, epilessia e disabilità intellettive. Sebbene non esista una cura per questa condizione, una nuova ricerca presso la UNC School of Medicine sta preparando il terreno per lo sviluppo di un trattamento.
Ben Philpot, Ph.D., Professore emerito di biologia cellulare e fisiologia presso la facoltà di medicina dell’UNC e Direttore associato dell’UNC Neuroscience Center e il suo laboratorio hanno identificato una piccola molecola che potrebbe essere sicura, somministrata in modo non invasivo e in grado di “attivare” la copia dormiente del gene UBE3A ereditato per via paterna in tutto il cervello, il che porterebbe al corretto funzionamento delle proteine e delle cellule, il che rappresenterebbe una sorta di terapia genica per gli individui affetti dalla sindrome di Angelman.
“Questo composto da noi identificato ha dimostrato di avere un assorbimento eccellente nei cervelli in via di sviluppo dei modelli animali”, ha affermato Philpot, uno dei massimi esperti della sindrome di Angelman. “Abbiamo ancora molto lavoro da fare prima di poter iniziare una sperimentazione clinica, ma questa piccola molecola fornisce un punto di partenza eccellente per sviluppare un trattamento sicuro ed efficace per la sindrome di Angelman“.
Questi risultati, pubblicati su Nature Communications, segnano una pietra miliare importante nel campo, secondo Mark Zylka, WR Kenan Jr. Distinguished Professor of Cell Biology and Physiology presso la UNC School of Medicine e Direttore dell’UNC Neuroscience Center. “Nessun altro composto di piccole molecole ha ancora mostrato una tale promessa per la sindrome di Angelman“, ha aggiunto.
A differenza di altri disturbi monogenici come la fibrosi cistica e l’anemia falciforme, la sindrome di Angelman ha un profilo genetico unico. I ricercatori hanno scoperto che i bambini affetti da queste condizioni sono privi della copia ereditata dalla madre del gene UBE3A, mentre la copia ereditata dal padre del gene UBE3A rimane dormiente nei neuroni, come accade negli individui neurotipici.
In genere, l‘UBE3A aiuta a regolare i livelli di proteine importanti; la mancanza di una copia funzionante provoca gravi interruzioni nello sviluppo del cervello.
Per ragioni non del tutto chiare, la copia paterna di UBE3A è normalmente “disattivata” nei neuroni di tutto il cervello. Quindi, quando la copia materna del gene UBE3A è mutata, ciò porta a una perdita della proteina UBE3A nel cervello. Philpot e altri ricercatori hanno teorizzato che l’attivazione della copia paterna di UBE3A potrebbe aiutare a curare la condizione.
Hanna Vihma, Ph.D., ricercatrice post-dottorato presso il laboratorio Philpot e prima autrice dello studio e i suoi colleghi, hanno esaminato più di 2.800 piccole molecole da una libreria chemiogenetica Pfizer per determinare se una di esse potesse attivare in modo efficace l’UBE3A paterno nei modelli murini affetti dalla sindrome di Angelman.
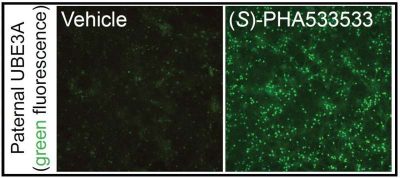
I ricercatori hanno modificato geneticamente le cellule neurali dei topi con una proteina fluorescente che si illumina quando viene attivato il gene paterno UBE3A. Dopo aver trattato i neuroni con oltre 2.800 piccole molecole per 72 ore, i ricercatori hanno confrontato le migliaia di cellule trattate con quelle trattate con topotecan, una piccola molecola nota che può attivare l’UBE3A paterno, ma che non aveva valore terapeutico nei modelli animali della condizione.
(S)-PHA533533, un composto precedentemente sviluppato come agente antitumorale, ha indotto i neuroni a esprimere una fluorescenza che rivaleggiava con quella indotta dal Topotecan, il che significa che il suo effetto era sufficientemente potente da attivare con successo l’UBE3A paterno.
I ricercatori sono stati in grado di confermare gli stessi risultati utilizzando cellule staminali pluripotenti indotte derivate da esseri umani affetti dalla sindrome di Angelman, il che indica che questo composto ha potenziale clinico.
Inoltre, i ricercatori hanno osservato che (S)-PHA533533 ha un’eccellente biodisponibilità nel cervello in via di sviluppo, il che significa che viaggia verso il suo bersaglio con facilità e rimane lì. Ciò è degno di nota in quanto le precedenti terapie genetiche per la sindrome di Angelman avevano una biodisponibilità più limitata.
“Abbiamo precedentemente dimostrato che il Topotecan, un inibitore della topoisomerasi, aveva una biodisponibilità molto scarsa nei modelli di topi”, ha affermato Vihma. “Siamo stati in grado di dimostrare che (S)-PHA533533 aveva un migliore assorbimento e che la stessa piccola molecola poteva essere tradotta in cellule neurali derivate dall’uomo, il che è una scoperta enorme. Ciò significa che, o un composto simile, ha un vero potenziale come trattamento per i bambini“.
Sebbene (S)-PHA533533 si mostri promettente, i ricercatori stanno ancora lavorando per identificare il bersaglio preciso all’interno delle cellule che causa gli effetti desiderati del farmaco. Philpot e colleghi devono anche condurre ulteriori studi per perfezionare la chimica medicinale del farmaco per garantire che il composto, o un’altra sua versione, sia sicuro ed efficace per un uso futuro in ambito clinico.
Leggi anche:Sindrome di Angelman: nuovo bersaglio farmacologico per un trattamento migliore
“È improbabile che questo sia esattamente il composto che porteremmo in clinica”, ha affermato Philpot. Insieme ai chimici farmaceutici del laboratorio di Jeff Aubé, Ph.D., il laboratorio Philpot sta lavorando per identificare molecole simili con proprietà farmacologiche e profili di sicurezza migliorati. “Tuttavia, questo ci fornisce un composto con cui possiamo lavorare per creare un composto ancora migliore che potrebbe essere portato in clinica”.
Fonte:Nature Communications