Sindrome dell’X fragile-Immagine: astratto grafico Credito:Cell (2023).
I ricercatori hanno scoperto nuovi geni alterati e un modello molecolare inaspettato, chiamato BREACH, correlato alla sindrome dell’X fragile (FXS), una malattia genetica che, secondo i Centri per il controllo e la prevenzione delle malattie, colpisce circa 1 maschio su 7.000 e circa 1 femmina su 11.000.
Lo studio, condotto da ricercatori della Perelman School of Medicine dell’Università della Pennsylvania, che ha utilizzato cellule e tessuto cerebrale donati dai pazienti, ha anche dimostrato che semplicemente modificando la lunghezza dello schema ripetitivo anomalo si potrebbero ripristinare i geni silenziati su più cromosomi.
Lo studio è stato pubblicato sulla rivista Cell.
“I nostri risultati hanno implicazioni per le future strategie di trattamento della sindrome dell’X fragile ed evidenziano potenziali meccanismi che contribuiscono all’instabilità del genoma che potrebbe essere alla base anche di altre malattie“, ha affermato la coautrice dello studio Linda Zhou, MD, Ph.D., della Dermatologia alla Penn Medicine.
Un team guidato dall’autrice senior Jennifer Phillips-Cremins, Ph.D., Professore associato di Bioingegneria e Genetica e membro dell’Epigenetics Institute della Penn Medicine, ha studiato l’FXS, la forma più comune di disabilità intellettiva ereditaria, al fine di giungere alla comprensione della causa sottostante del disturbo. I modelli da manuale la attribuiscono al silenziamento di un singolo gene, FMR1, e alla perdita della proteina codificata da FMR1, la ribonucleoproteina Fragile X Messenger (FMRP).
È opinione diffusa che la perdita di FMRP causi una grave disregolazione delle sinapsi, che collegano i neuroni nel cervello, nonché l’interruzione del modo in cui i geni vengono espressi nei nuclei dei neuroni.
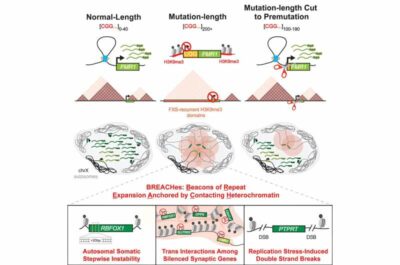
Il modello principale di FXS è stato costruito su studi condotti su topi transgenici in cui il gene FMR1 era stato eliminato. Tuttavia, al modello murino mancava il driver genetico critico di FXS: una mutazione chiamata “espansione di ripetizione”, che si verifica quando una lunga ripetizione di una sequenza di due o più lettere di DNA diventa instabile e anormalmente lunga (una ripetizione della lunghezza della mutazione).
Per FXS, questa è la sequenza di tre lettere, CGG, che appare a un’estremità del gene FMR1. Mentre una versione normale di FMR1 ha 40 o meno triplette CGG nel tratto ripetuto, un paziente FXS avrà 200 triplette o più. L’anomalia innesca una risposta difensiva da parte della cellula, che sostanzialmente mette a tacere FMR1 e FMRP. Poiché la sequenza ripetitiva è difficile da progettare, i modelli animali di piccola taglia di FXS mancano del tratto ripetuto e quindi potrebbero non aver dimostrato aspetti importanti del ruolo del DNA ripetitivo nei meccanismi alla base di FXS.
Nel nuovo studio, il gruppo di ricerca ha utilizzato una serie di tecniche avanzate di sequenziamento e imaging, nonché linee cellulari umane e tessuto cerebrale con l’espansione ripetuta CGG, per scoprire nuovi sorprendenti modelli di distruzione del genoma in FXS. I ricercatori hanno scoperto che ampie fasce di cromosomi multipli nei campioni di pazienti FXS, che includono la ripetizione CGG, sono contrassegnate con l’eterocromatina silenziante. Questi “domini” dell’eterocromatina sono chiamati BREACH, segnali di espansione ripetuta, ancoraggio e contatto con l’eterocromatina.
Le “violazioni” si uniscono in gruppi che entrano in contatto fisico nel nucleo e silenziano i geni coinvolti nelle connessioni sinaptiche dei neuroni, insieme ai geni legati all’integrità del tessuto connettivo come la pelle e le articolazioni. Nei pazienti FXS in clinica si osservano interruzioni delle sinapsi e del tessuto connettivo, pertanto la capacità di identificare i BREACH ha la possibilità di essere un potente strumento per trovare geni interrotti potenzialmente importanti oltre a FMR1.
I ricercatori hanno anche testato se la ripetizione potesse essere direttamente collegata ai BREACH utilizzando la tecnologia di modifica genetica CRISPR-Cas per ridurre l’espansione CGG a una lunghezza che non causa FXS.
“Quando abbiamo tagliato il CGG a una lunghezza più breve chiamata premutazione (100-190 triplette), abbiamo osservato che molte delle ampie fasce di silenziamento dell’eterocromatina erano invertite e che più cromosomi erano spazialmente disconnessi da FMR1“, ha affermato il co-autore principale Ken Chandradoss, Ph.D. e Ravi Boya, Ph.D., entrambi ricercatori post-dottorato nel laboratorio di Phillips-Cremins.
Gli esperimenti del team hanno dimostrato che i geni originariamente silenziati dai BREACH venivano riespressi nelle cellule FXS con la ripetizione CGG accorciata da CRISPR.
“L’ampio impatto della nostra scoperta che l’espansione CGG della lunghezza della mutazione è necessaria per il mantenimento dei BREACH è che la ripetizione da sola può essere potenzialmente utilizzata come approccio terapeutico per invertire il silenziamento dell’intero genoma di più geni critici che potenzialmente contribuiscono alle presentazioni cliniche FXS “, ha detto il co-autore principale Thomas Malachowski, un Ph.D. studente nel laboratorio di Cremins.
I futuri trattamenti FXS potrebbero esplorare la sostituzione delle funzioni di alcuni dei geni silenziati identificati nello studio, non solo di FMR1. I ricercatori hanno notato, tuttavia, che una strategia più ambiziosa sarebbe quella di ridurre l’espansione delle ripetizioni CGG eccessivamente lunghe in un momento definito nello sviluppo per prevenire o almeno invertire gli effetti del silenziamento dell’eterocromatina.
Per esplorare questa possibilità sarebbe necessario bilanciare attentamente gli effetti positivi della riattivazione di geni importanti con il ruolo protettivo che l’eterocromatina ha nella protezione dall’instabilità del genoma ripetitivo.
Esempi di altri disturbi potenzialmente influenzati da questi risultati includono la malattia di Huntington e la sclerosi laterale amiotrofica (malattia di Lou Gehrig), che sono membri della stessa, più ampia classe di disturbi ad espansione ripetuta come FXS, che si ritiene siano guidati dalla mutazione di un singolo tratto ripetitivo del DNA.
Phillips-Cremins ha anche spiegato che il team ha osservato “violazioni” in altri modelli cellulari umani di instabilità del genoma, comprese linee cellulari contenenti mutazioni riscontrate nel cancro o rotture del DNA indotte in laboratorio.
Leggi anche:Sindrome dell’X Fragile: cambiamento significativo nella comprensione
“I nostri risultati suggeriscono che in futuro si potrebbe scoprire che i BREACH hanno un impatto più ampio sul silenziamento genico in altre malattie con instabilità del genoma, inclusi alcuni tumori e altri disturbi dell’espansione ripetuta“, ha affermato Cremins.
Fonte:Cell