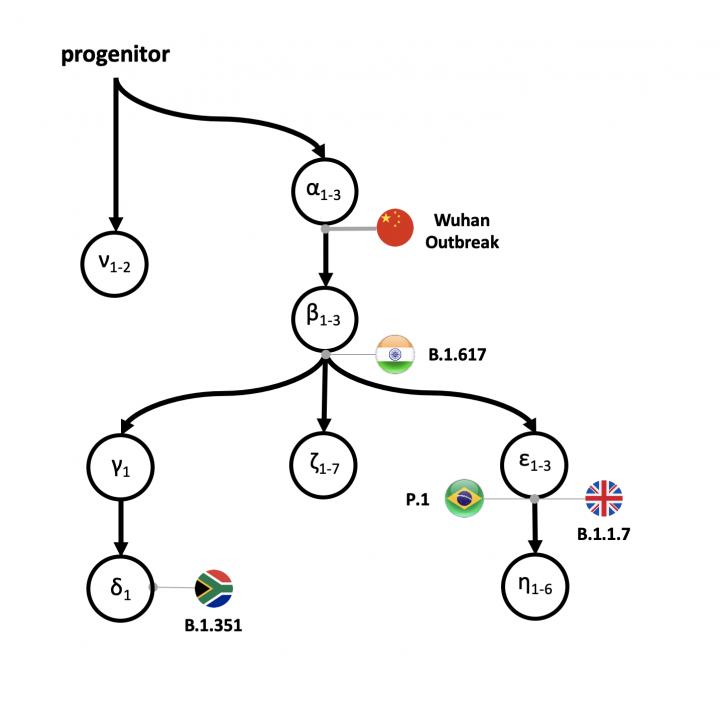
(SARS-CoV-2-Immagine:il virus progenitore (proCoV2) e i suoi discendenti iniziali sono nati in Cina, sulla base delle prime mutazioni di proCoV2 e delle loro posizioni, che sono state ricondotte a verificarsi 6-8 settimane prima dell’epidemia di Wuhan in Cina. Inoltre, il team scientifico ha anche dimostrato che una popolazione di ceppi con almeno tre differenze mutazionali (alfa 1-3) da proCoV2 esisteva al momento del primo rilevamento di casi di COVID-19 in Cina. Credit:Sudhir Kumar, Temple University).
Vedi anche:SARS-CoV-2: cosa succede nei primi giorni di infezione?
Nel campo dell’epidemiologia molecolare, la comunità scientifica mondiale ha costantemente indagato per risolvere l’enigma della storia primitiva di SARS-CoV-2. Nonostante i recenti sforzi dell’Organizzazione Mondiale della Sanità, nessuno fino ad oggi ha identificato il primo caso di trasmissione umana, o “paziente zero”, nella pandemia COVID-19.
È necessario trovare il primo caso possibile per capire meglio come il virus possa essere saltato nel suo ospite animale prima di infettare gli esseri umani, nonché la storia di come il genoma virale SARS-CoV-2 sia mutato nel tempo e si sia diffuso a livello globale.
Da quando la prima infezione da virus SARS-CoV-2 è stata rilevata nel dicembre 2019, ben oltre un milione di genomi di SARS-CoV-2 sono stati sequenziati in tutto il mondo, rivelando che il coronavirus sta mutando, anche se lentamente, a una velocità di 25 mutazioni per genoma per anno. L’enorme numero di varianti emergenti, tra cui quella del Regno Unito (B.1.1.1.7), sudafricana (B.1.351), sudamericana (P.1) e ora indiana (B.1.617) non solo hanno sostituito i precedente dominanti ceppi nelle rispettive regioni, ma minacciano ancora la salute mondiale a causa del loro potenziale di sfuggire ai vaccini e alle terapie di oggi.
“Il virus SARS-CoV-2 ha già infettato più di 145 milioni di persone e causato 3 milioni di morti in tutto il mondo”, ha detto Sudhir Kumar, Direttore dell’Istituto di genomica e medicina evolutiva alla Temple University. “Abbiamo deciso di trovare l’antenato comune genetico di tutte queste infezioni, che chiamiamo genoma progenitore“. Questo genoma progenitore (proCoV2) è la madre di tutti i coronavirus SARS-CoV-2 che hanno infettato e continuano a infettare le persone oggi.
Spiegano gli autori:
“Il sequenziamento globale di centinaia di migliaia di genomi della sindrome respiratoria acuta grave coronavirus 2, SARS-CoV-2, ha continuato a rivelare nuove varianti genetiche che sono la chiave per svelare la sua prima storia evolutiva e monitorare la sua diffusione globale nel tempo. Qui, presentiamo la storia mutazionale criptica finora e le dinamiche spaziotemporali di SARS-CoV-2 da un’analisi di migliaia di genomi di alta qualità. Riportiamo il probabile antenato comune più recente di SARS-CoV-2, ricostruito attraverso una nuova applicazione e il progresso di metodi computazionali inizialmente sviluppati per inferire la storia mutazionale delle cellule tumorali in un paziente. Questo genoma progenitore differisce dai genomi dei primi coronavirus campionati in Cina per tre varianti, il che implica che nessuno dei primi pazienti rappresenta il caso indice o ha dato origine a tutte le infezioni umane. Tuttavia, più infezioni da coronavirus in Cina e negli Stati Uniti hanno ospitato l’impronta genetica del progenitore nel gennaio 2020 e successivamente, suggerendo che il progenitore si stava diffondendo in tutto il mondo mesi prima e dopo i primi casi segnalati di COVID-19 in Cina. Le mutazioni del progenitore e dei suoi derivati hanno prodotto molti ceppi dominanti di coronavirus, che si sono diffusi episodicamente nel tempo. Il fingerprinting basato su mutazioni comuni rivela che la stessa linea di coronavirus ha dominato il Nord America per la maggior parte della pandemia nel 2020. Ci sono state sostituzioni multiple di ceppi di coronavirus predominanti in Europa e Asia e la continua presenza di più ceppi ad alta frequenza in Asia e Nord America“.
Il gruppo di Kumar stima che il progenitore SARS-CoV-2 stesse già circolando con una linea temporale precedente, almeno 6-8 settimane prima del primo genoma sequenziato in Cina, noto come Wuhan-1. “Questa sequenza temporale indica la presenza di proCoV2 alla fine di ottobre 2019, il che è coerente con il rapporto di un frammento di proteina spike identica a Wuhan 1 all’inizio di dicembre in Italia, tra le altre prove”, ha detto Sayaka Miura, autore senior dello studio.
“Abbiamo trovato l’impronta genetica del progenitore nel gennaio 2020 e successivamente in più infezioni da coronavirus in Cina e negli Stati Uniti. Il progenitore si stava diffondendo in tutto il mondo mesi prima e dopo i primi casi segnalati di COVID-19 in Cina”, ha detto Pond. Oltre alle loro scoperte sulla storia iniziale di SARS-CoV-2, il gruppo di Kumar ha anche sviluppato impronte mutazionali intuitive e classificazione dei simboli greci (ν, α, β, γ, δ e ε) per semplificare la categorizzazione dei principali ceppi, sub-ceppi e varianti che infettano un individuo o colonizzano una regione globale. Ciò può aiutare gli scienziati a tracciare meglio e fornire un contesto per l’ordine di comparsa di nuove varianti.
“Nel complesso, le nostre impronte digitali mutazionali e la nomenclatura forniscono un modo semplice per raccogliere l’ascendenza di nuove varianti rispetto alle designazioni filogenetiche, ad esempio, B.1.351 e B.1.1.7”, ha detto Kumar. Ad esempio, un’impronta digitale α si riferisce ai genomi con una o più delle varianti α e nessun’altra variante principale successiva, e un’impronta digitale αβ si riferisce ai genomi che contengono tutte le varianti α, almeno una variante β e nessun’altra variante principale. “Con i nostri strumenti, abbiamo osservato la diffusione e la sostituzione dei ceppi prevalenti in Europa (αβε con αβζ) e in Asia (α con αβε), la preponderanza dello stesso ceppo per la maggior parte della pandemia in Nord America (αβ? Δ) e la continua presenza di più ceppi ad alta frequenza in Asia e Nord America “, ha spiegato Pond.
Arrivare alla radice del problema
Per identificare il genoma progenitore, i ricercatori hanno utilizzato un approccio non applicato in precedenza a SARS-CoV-2, chiamato analisi dell’ordine di mutazione. La tecnica, ampiamente utilizzata nella ricerca sul cancro, si basa su un’analisi clonale dei ceppi mutanti e sulla frequenza con cui le coppie di mutazioni appaiono insieme per trovare la radice del virus.
“Molti precedenti tentativi di analizzare set di dati così grandi non hanno avuto successo a causa della concentrazione sulla costruzione di un albero evolutivo di SARS-CoV-2”, afferma Kumar. “Questo coronavirus si evolve troppo lentamente, il numero di genomi da analizzare è troppo grande e la qualità dei dati dei genomi è molto variabile. Ho immediatamente visto parallelismi tra le proprietà di questi dati genetici del coronavirus con i dati genetici della diffusione clonale di un’altra malattia nefasta, il cancro”.
Kumar e Miura hanno sviluppato e studiato molte tecniche per analizzare i dati genetici dei tumori nei malati di cancro. Hanno adattato e innovato queste tecniche per costruire una scia di mutazioni che risalivano all’impronta genetica del progenitore. “L’approccio di rilevamento della mutazione ha prodotto il progenitore e la storia familiare della sua mutazione principale. È un ottimo esempio di come i big data accoppiati con il data mining biologicamente informato rivelano modelli importanti”, ha detto Kumar.
Emerge una cronologia precedente. “Questo genoma progenitore aveva una sequenza molto diversa da quella che alcune persone chiamano sequenza di riferimento, che è quella che è stata osservata per prima in Cina e depositata nel database GISAID SARS-CoV-2“, ha detto Kumar.
La corrispondenza più vicina era quella di otto genomi campionati da 26 a 80 giorni dopo il primo virus campionato dal 24 dicembre 2019. Sono state trovate più corrispondenze ravvicinate in tutti i continenti campionati e rilevate fino a giugno 2020 (giorno 181 della pandemia) in Sud America. Complessivamente, 140 genomi del gruppo di Kumar analizzati contenevano tutti solo differenze sinonime da proCoV2. Cioè, tutte le loro proteine erano identiche alle corrispondenti proteine proCoV2 nella sequenza amminoacidica. La maggioranza (93 genomi) di queste corrispondenze a livello di proteine proveniva da coronavirus campionati in Cina e in altri paesi asiatici.
Questi modelli spaziotemporali hanno suggerito che proCoV2 possedeva già l’intero repertorio di sequenze proteiche necessarie per infettare, diffondersi e persistere nella popolazione umana globale.
I ricercatori hanno scoperto che il virus proCoV2 e i suoi discendenti iniziali sono nati in Cina, sulla base delle prime mutazioni di proCoV2 e delle loro posizioni. Inoltre, hanno anche dimostrato che una popolazione di ceppi con almeno tre differenze mutazionali da proCoV2 esisteva al momento del primo rilevamento di casi di COVID-19 in Cina. Con stime di SARS-CoV-2 che acquisiscono 25 mutazioni all’anno, il virus deve aver già infettato le persone diverse settimane prima dei casi di dicembre 2019.
Firme mutazionali
Poiché c’erano forti prove di molte mutazioni prima di quelle trovate nel genoma di riferimento, il gruppo di Kumar ha dovuto elaborare una nuova nomenclatura delle firme mutazionali per classificare SARS-CoV-2 e spiegarle introducendo una serie di simboli di lettere greche. Ad esempio, i ricercatori hanno scoperto che l’emergere di varianti del genoma α SARS-CoV-2 era anteriore alle prime segnalazioni di COVID-19. Ciò implica fortemente l’esistenza di una certa diversità di sequenza nelle popolazioni ancestrali SARS-CoV-2. Tutti i 17 genomi campionati dalla Cina nel dicembre 2019, incluso il genoma di riferimento SARS-CoV-2 designato, portano tutte e tre le varianti α. Tuttavia, 1.756 genomi senza varianti α sono stati campionati in tutto il mondo fino a luglio 2020. Pertanto, i primi genomi campionati (incluso il riferimento designato) non erano i ceppi progenitori. Prevede anche che il genoma progenitore avesse una prole che si stava diffondendo in tutto il mondo durante le prime fasi di COVID 19. SARS-CoV-2 era pronto a infettare fin dall’inizio.
“Il progenitore aveva tutte le capacità di cui aveva bisogno per diffondersi”, ha detto Pond. “C’è una sovrabbondanza di cambiamenti non sinonimi nella popolazione. Quello che è successo tra pipistrelli e umani rimane poco chiaro, ma proCoV2 potrebbe già infettare su scala pandemica”.
Una diffusione globale
Complessivamente, i ricercatori hanno identificato sette principali lignaggi evolutivi e la natura episodica della loro diffusione globale. Il genoma proCoV2 ha dato origine a molte delle principali linee di discendenza, alcune delle quali sono nate in Europa e Nord America dopo la probabile genesi delle linee ancestrali in Cina. “I ceppi asiatici hanno fondato l’intera pandemia”, ha detto Kumar. “Ma nel tempo, molte varianti che si sono evolute altrove stanno ora infettando l’Asia molto di più”.
Le loro analisi mutazionali hanno anche stabilito che i coronavirus nordamericani ospitano firme genomiche molto diverse da quelle prevalenti in Europa e in Asia.
“Questo è un processo dinamico”, ha detto Kumar. “Chiaramente, ci sono immagini molto diverse di diffusione dipinte dall’emergere di nuove mutazioni, i tre ε, γ e delta, che abbiamo scoperto che si verificano dopo il cambiamento della proteina spike (una mutazione β). Gli scienziati stanno ancora cercando di capire se le proprietà di queste mutazioni hanno accelerato la pandemia”.
Sorprendentemente, la firma mutazionale di αβ Δ è rimasta la stirpe dominante in Nord America dall’aprile 2020, in contrasto con il turn over visto in Europa e in Asia. Più recentemente, nuove varianti a rapida diffusione, inclusa una variante della proteina S (N501Y) dal Sud Africa e dal Regno Unito (B.1.1.17), sono aumentate rapidamente. I coronavirus con variante N501Y in Sud Africa portano l’impronta genetica αβγδ, mentre quelli nel Regno Unito portano l’impronta genetica αβε, secondo il loro schema di classificazione. “Pertanto, l’antenato αβ continua a dare origine a molte delle principali propaggini di questo coronavirus“, ha detto Kumar.
Aggiornamenti in tempo reale
Lo studio MBE si basava su tre istantanee recuperate da GISAID il 7 luglio 2020 (un set di dati di 60.332 genomi), il 12 ottobre 2020 (conteneva 133.741 genomi) e, infine, un set di dati ampliato di 172.480 genomi campionati il 30 dicembre, 2020.
Andando avanti, i ricercatori continueranno a perfezionare i risultati non appena saranno disponibili nuovi dati.
“Più di un milione di genomi di SARS-CoV-2 sono sequenziati ora”, ha detto Pond. “Il potere di questo approccio è che più dati hai, più facilmente puoi dire la frequenza precisa delle singole mutazioni e coppie di mutazioni. Queste varianti prodotte, le varianti a singolo nucleotide o SNV, la loro frequenza e la cronologia possono essere raccontato molto bene con più dati. Pertanto, le nostre analisi deducono una radice credibile per la filogenesi SARS-CoV-2”.
Lo studio MBE fa parte del loro sforzo per mantenere un monitoraggio continuo e in tempo reale dei genomi SARS-CoV-2 che ora sono cresciuti fino a includere oltre 350.000 genomi .
“Abbiamo creato una dashboard in tempo reale che mostra i risultati regolarmente aggiornati perché i processi di analisi dei dati, preparazione del manoscritto e revisione tra pari di articoli scientifici sono molto più lenti del ritmo di espansione della raccolta del genoma di SARS-CoV-2”, ha detto Pond. “Forniamo anche un semplice strumento “browser” per classificare qualsiasi genoma di SARS-CoV-2 in base alle mutazioni chiave derivate dall’analisi MOA. Questi risultati e le nostre intuitive impronte digitali mutazionali e codici a barre dei ceppi SARS-CoV-2 hanno superato sfide scoraggianti per sviluppare una retrospettiva su come, quando e perché COVID-19 è emerso e si è diffuso, che è un prerequisito per la creazione di rimedi per superare questa pandemia attraverso gli sforzi della scienza, della tecnologia, delle politiche pubbliche e della medicina “, ha concluso Kumar.
Fonte:Oxford Academic