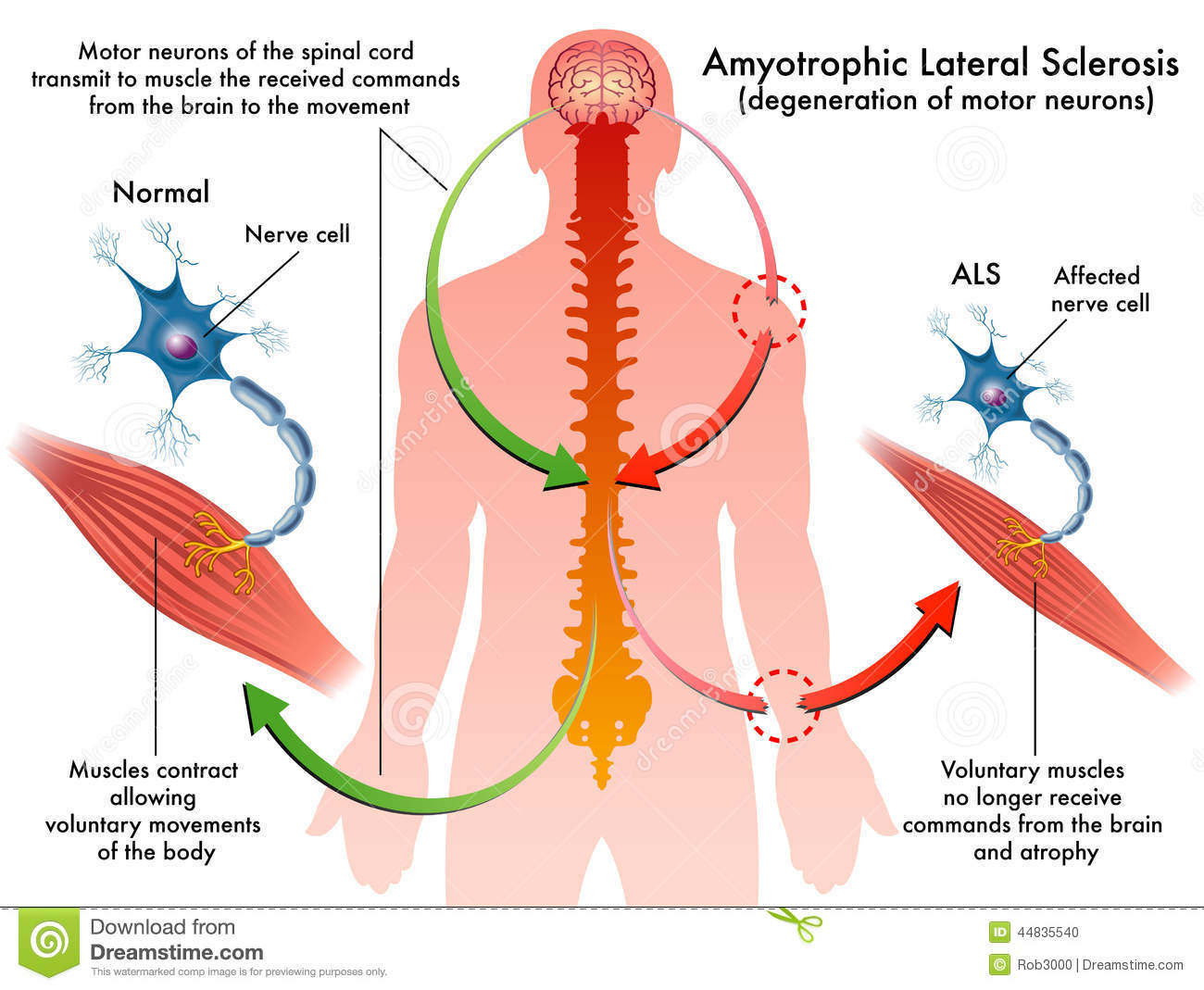
“La Sclerosi Laterale Amiotrofica (SLA), conosciuta anche come “Morbo di Lou Gehrig”,” malattia di Charcot” o “malattia del motoneurone”, è una malattia neurodegenerativa progressiva che colpisce i motoneuroni, cioè le cellule nervose cerebrali e del midollo spinale che permettono i movimenti della muscolatura volontaria. Esistono due gruppi di motoneuroni; il primo (primo motoneurone o motoneurone corticale) si trova nella corteccia cerebrale e trasporta il segnale nervoso attraverso prolungamenti che dal cervello arrivano al midollo spinale. Il secondo (2° motoneurone) è invece formato da cellule nervose che trasportano il segnale dal tronco encefalico e dal midollo spinale ai muscoli. La SLA è caratterizzata dal fatto che sia il primo che il secondo motoneurone vanno incontro a degenerazione e muoiono. La morte di queste cellule avviene gradualmente nel corso di mesi o anche anni. Tra le possibili cause c’è l’accumulo di proteine anomale all’interno della cellula: l’accumulo di proteine alterate all’interno del motoneurone contribuisce a portare la cellula alla morte. Nella maggior parte dei pazienti la proteina che si accumula è denominata TDP43 (sia nelle forme sporadiche che in alcune forme famigliari); nei pazienti portatori di mutazione del gene SOD1 la proteina anomala è la SOD1 (codificata dal gene alterato) e nei pazienti con mutazione del gene FUS è la proteina omonima“.
I ricercatori negli ultimi anni hanno concentrato la loro attenzione sulle proteine malfunzionanti responsabili della morte dei neuroni nel cervello e nel midollo spinale. In un nuovo studio pubblicato sulla rivista Structure, i ricercatori della Facoltà di Medicina UNC hanno annunciato per la prima volta, che la stabilizzazione di una proteina chiamata SOD1 può contribuire a invertire il processo di accumulo di aggregati tossici nei neuroni colpiti dalla Sclerosi Laterale Amiotrofica (SLA).
Attualmente la SLA non ha cura e le sue cause rimangono in gran parte misteriose.
Oltre a mostrare che la stabilizzazione SOD1 è protettiva per le cellule simili ai neuroni motori, il nuovo studio è anche il primo ad aver trovato un modo per stabilizzare SOD1, offrendo nuovi interessanti spunti per la ricerca di farmaci che potrebbero potenzialmente impedire la malattia o rallentare la sua progressione.
“Attraverso un processo naturale chiamato fosforilazione, si può stabilizzare SOD1 nelle cellule e prevenire la formazione di oligomeri tossici nelle persone senza malattia”, ha detto l’autore senior dello studio Nikolay Dokholyan che ha condotto lo studio in collaborazione con Michael Hooker, Prof. di Biochimica e Biofisica presso la UNC. “La comprensione dei meccanismi cellulari con conseguente fosforilazione SOD1 non solo offre approfondimenti su come le cellule rispondono agli aggregati tossici di SOD1, ma offre intuizioni nuove per lo sviluppo di strategie farmaceutiche volte a promuovere la fosforilazione di SOD1. Questo è il nostro obiettivo immediato”, ha aggiunto il ricercatore.
I pazienti affetti da SLA soffrono di paralisi progressiva e morte precoce a causa della perdita di neuroni motori che sono cruciali per muoversi, parlare, deglutire e respirare. In uno studio pubblicato lo scorso anno, la squadra di Dokholyan ha scoperto che nel caso di mutazione del gene SOD1, la proteina SOD si presenta sotto forma di “trimeri” in grado di uccidere le cellule simili ai neuroni motori, come risultato dalle sperimentazioni condotte in laboratorio. Quando funziona correttamente, SOD1 esiste in strutture note come “dimeri”.
“L’idea era che se siamo in grado di stabilizzare SOD1, in primo luogo, siamo in grado potenzialmente di fornire un modo per prevenire la malattia in una fase iniziale”, ha detto Cheng Zhu, ricercatore post-dottorato nel laboratorio di Dokholyan e autore del nuovo studio . “I nostri risultati mostrano che la stabilizzazione di SOD1 può aumentare la vitalità delle cellule”.
I risultati potrebbero essere particolarmente rilevanti per un sottoinsieme di casi di SLA – circa 1/2 per cento – che sono associati con variazioni nella proteina SOD1. Tuttavia, SOD1 è implicata nella formazione di aggregati tossici anche in pazienti senza mutazioni nei loro geni SOD1, suggerendo che la stabilizzazione della proteina potrebbe beneficiare molti altri pazienti.
Poichè trimeri SOD1 si disintegrano quasi non appena si formano, il team di ricerca ha prima utilizzato la modellazione computazionale per fare ipotesi plausibili su quali tipi di modifiche si potrebbero apportare a SOD1 per evitare che causi la formazione di aggregati tossici. I loro modelli suggeriscono che l’aggiunta di un gruppo fosfato, noto come fosforilazione, potrebbe aiutare a stabilizzare la proteina. La fosforilazione è nota per influenzare il funzionamento delle proteine nel contesto di molti processi cellulari.
Per verificare questa ipotesi, il team ha creato una mutazione genetica che imitava l’aggiunta di un gruppo fosfato a proteine SOD1 in cellule simili ai neuroni motori coltivati in laboratorio. Le cellule in coltura usate sono state sviluppate come parte del precedente lavoro per modellare i meccanismi alla base della SLA e presentavano già una mutazione che aveva indotto le proteine SOD1 a formare grumi tossici.
“Quando abbiamo trasferito questa nuova mutazione nelle cellule, di concerto con la mutazione che causa la malattia, in realtà le abbiamo salvate dalla tossicità e impedito la loro morte”, ha spiegato Jimmy Fay, uno studente laureato presso la UNC ed ex tecnico di laboratorio che ha preso parte alla ricerca. Invece di essere uccise dai ciuffi tossici di SOD1, le cellule sono sopravvissute grazie alla mutazione che imita la fosforilazione.
I risultati offrono due nuove strade per individuare possibili bersagli farmacologici per il trattamento della SLA. Una è quella di trovare il modo di promuovere la fosforilazione di SOD1 nei neuroni motori del paziente, l’altra è quella di cercare altri modi per stabilizzare SOD1 che tende ad aggregarsi.
“Ora possiamo vedere una via d’uscita”, ha detto Fay. “Sappiamo che questa mutazione stabilizza SOD1 e la speranza è che possiamo trovare un farmaco che pianifica attraverso questo processo, il comportamento della proteina”.
I risultati possono anche fornire indizi sul perché alcune persone si ammalano di SLA, mentre altre no. Se la fosforilazione di SOD1 si trova ad essere comune nelle persone senza SLA, si potrebbe pensare che i difetti che portano alla ridotta fosforilazione svolgono un ruolo nella destabilizzazione di SOD1 anche nelle persone senza mutazioni SOD1 dannose.
Fonte: Structure