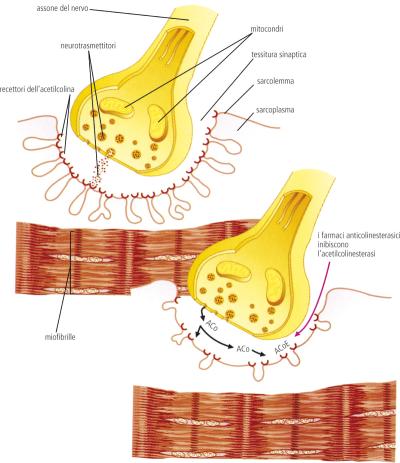
Anche se sono stati compiuti progressi significativi negli ultimi 25 anni per identificare anomalie genetiche associate alla sindrome miastenica congenita (CMS), molti pazienti rimangono geneticamente non diagnosticati.
Un rapporto, pubblicato nel numero inaugurale del Giornale delle Malattie Neuromuscolari, identifica un difetto genetico nei mitocondri, in particolare in SLC25A1 portante citrato, che può essere alla base del deficit nella trasmissione neuromuscolare.
“Mentre difetti genetici mitocondriali possono causare una miriade di disturbi neurologici, tra cui miopatie e neuropatie, questi non sono stati specificamente implicati nei difetti della giunzione neuromuscolare”, afferma Hanns lochmüller, MD, Professore dell’ Experimental Myology, Institute of Genetic Medicine, MRC Centre for Neuromuscular Diseases, Newcastle University, Newcastle upon Tyne, UK.
Dei 19 geni che sono stati implicati nella miastenia congenita, la maggior parte esprime proteine coinvolte nello sviluppo elle sinapsi neuromuscolari e funzione. Queste mutazioni coinvolgono di solito le proteine post-sinaptiche.
Lo studio attuale sposta l’area di danno alla regione presinaptica.
I ricercatori hanno condotto analisi genomiche su due pazienti che sono fratello e sorella. La coppia è nata da genitori sani che erano cugini di primo grado. “La storia della famiglia era altamente suscettibile di trasmissione autosomica recessiva”, osserva il dottor lochmüller. Fin da bambino, uno dei fratelli ormai di 33 anni, aveva mostrato problemi motori che peggioravano con l’esercizio e miglioravano con il riposo. Il giovane aveva una lieve ptosi bilaterale (abbassamento della palpebra), difficoltà di parola e difficoltà di apprendimento lievi. Sua sorella di 19 anni, ha mostrato un ritardo di sviluppo, cadute ricorrenti, debolezza degli arti, intermittente visione doppia e abbassamento dei muscoli facciali.
I ricercatori hanno eseguito la mappatura omozigosi e l’intero sequenziamento exome per determinare la causa genetica di fondo della condizione dei due fratelli “e hanno identificato con successo una mutazione omozigote nel gene SLC25A1 che è un vettore citrato mitocondriale ritenuto una componente chiave in molti importanti processi biologici, come acidi grassi e biosintesi degli steroli, la gluconeogenesi, la glicolisi, il mantenimento dell’integrità cromosomica e la regolazione di autofagia.
Utilizzando tecniche elettrofisiologiche, i ricercatori sono stati in grado di mostrare le anomalie evidenti nelle giunzioni neuromuscolari dei pazienti.
I ricercatori hanno anche trovato la prova che SLC25A1 è necessario per la normale formazione della giunzione neuromuscolare, cercando gli effetti della ridotta espressione di SLC25A1 in embrioni di zebrafish. Anatomicamente, mentre le fibre muscolari apparivano normali, i terminali degli assoni motori presinaptici si presentavano accorciati e cresciuti in modo irregolare, senza evidenza di formazione di sinapsi complete. Il team ha anche notato cambiamenti strutturali nel cervello e nel cuore, che rispecchiavano anomalie osservate negli esseri umani.
Se un paziente mostra debolezza, può essere opportuno verificare le mutazioni SLC25A1 e considerare lo screening per difetti cardiaci e metabolici per trovare queste mutazioni.
“Abbiamo puntato a identificare il difetto molecolare alla base di questa famiglia”, aggiunge il co-ricercatore Kate Bushby, dell’ Università di Newcastle.”Siamo lieti che la più recente tecnologia di sequenziamento ha risolto questo puzzle diagnostico di lunga data, che ci aiuta nella consulenza e trattamento di questa condizione”.
La sindromi congenita miastenica (CMS) rappresenta un gruppo di malattie neuromuscolari ereditarie caratterizzate da debolezza muscolare (miastenia). I sintomi tipici sono la debolezza dei muscoli che controllano gli arti, come pure di quelli che sono coinvolti nel controllo degli occhi, la respirazione e movimenti del viso, della testa e del collo (a causa del coinvolgimento del tratto corticobulbare). I sintomi sono affaticamento, debolezza e la gravità dei deficit possono variare da lievi a gravi.
Fonte Mutations in the Mitochondrial Citrate Carrier SLC25A1 Are Associated with Impaired Neuromuscular Transmission.Journal of Neuromusclar Diseases, Volume 1/Issue 1 (June 2014) DOI:10.3233/JND-140021