Piccoli scostamenti nelle cellule del corpo a volte possono avere gravi conseguenze. I ricercatori di Berlino hanno scoperto perché le cellule dei pazienti affetti dalla rara malattia muscolare miopatia miotubolare non possono funzionare correttamente. Attraverso il lavoro pubblicato su Nature, è diventato chiaro come un processo cellulare dinamico essenziale per lo sviluppo muscolare e la funzione cellulare è regolato da piccoli cambiamenti di alcuni lipidi di membrana.
Se un bambino nasce con la miopatia miotubulare, la forma più grave di miopatie centronucleare (chiamata anche XLCNM), è appena in grado di respirare autonomamente.
Il gruppo di Volker Haucke dall’Istituto Leibniz di farmacologia molecolare (FMP) e Freie Universität (FUB) a Berlino, hanno condotto la ricerca in collaborazione con Jocelyn Laporte dell’ Institut Génétique Biologie Moléculaire Cellulaire (IGBMC) a Strasburgo e con Carsten Schultz dell’ European Molecular Biology Laboratory (EMBL) a Heidelberg ed hanno scoperto cosa va storto in questa malattia a livello molecolare ossia, un principio organizzativo generale nelle cellule.
Fino ad ora, è noto che questa malattia ereditaria comporta un difetto nel gene MTM1, a seguito del quale le fibre muscolari non funzionano normalmente. Il gene codifica per un enzima che è specializzato nella scissione di gruppi fosfato dai responsabili di alcuni lipidi di membrana chiamati fosfoinositide fosfati (PIP), ma come questo porta alla malattia era ancora sconosciuto.
PIP sono utilizzati dalla cellula per contrassegnare i suoi scomparti e per regolare il trasporto di sostanze. “La cellula è un sistema molto dinamico, che si può immaginare come una metropoli in cui le persone si muovono avanti e indietro”, spiega Volker Haucke. “A seconda delle occasioni, le persone cambiano i loro vestiti – se si indossa un cappotto o un abito, in una certa misura si assume un’identità diversa rispetto a quando si indossano jeans e felpa. In modo simile, gli scomparti e vescicole di trasporto di sostanze all’interno delle cellule utilizzano costantemente diversi PIP e quindi cambiano la loro identità “.
Ogni PIP consiste in una coda liposolubile che è ancorata nelle membrane dei compartimenti cellulari e una testa idrosolubile che sporge dalla membrana. La testa può essere caricata con fosfati in siti differenti, i gruppi fosfato sono staccati dagli enzimi e fissati in altri siti. Questo è un minimo cambiamento che avviene in un lampo, tuttavia è inequivocabilmente letto dalla cellula. Così, per esempio, se un gruppo fosfato è in una certa posizione, è chiaro che un contenitore di trasporto di sostanze è previsto per essere trasportato verso l’interno della cellula; se il tag fosfato è diverso, migra alla membrana cellulare esterna e scarica il suo trasporto all’esterno.
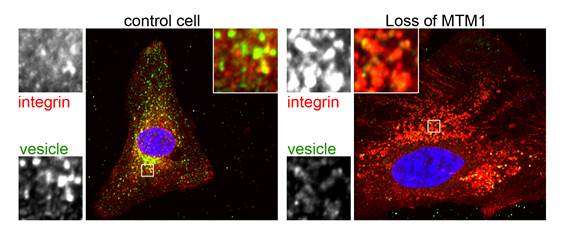
Questo tipo di trasporto è bloccato nei pazienti XLCNM, come è stato dimostrato da Katharina Ketel del gruppo Haucke, con esperimenti intricanti e immagini ad alta risoluzione dell’interno della cellula. La causa della malattia è un difetto di MTM1, un enzima che rimuove gruppi fosfato dai PIP e funziona solo in collaborazione con un altro enzima che attribuisce un gruppo fosfato ad un altro sito sulla testa idrosolubile di PIP. Questo chiarisce come i processi dinamici sono diretti nelle cellule e illustra come studiare una rara malattia genetica può scoprire un meccanismo molecolare essenziale per le nostre cellule, per funzionare correttamente.
“Nelle cellule sane, gruppi fosfato non vengono mai rimossi in modo casuale da PIP, perché un compartimento cellulare sarebbe poi improvvisamente lasciato completamente senza identità – che sarebbe l’ equivalente di una perdita di memoria”, spiega Volker Haucke. “Con l’aggiunta di PIP sintetici con un certo codice, siamo stati in grado di alterare il trasporto di sostanze a dimostrazione che la conversione del dell’ identità di PIP è realmente il problema delle cellule dei pazienti XLCNM”, aggiunge Castern Shultz.
“Nei pazienti XLCNM, alcuni dei contenitori di trasporto di sostanze da trasmettere originariamente alla superficie cellulare, restano bloccati all’interno della cellula perché un gruppo fosfato non può essere rimosso da un certo PIP” dice Jocelyn Laporte, un esperto in XLCNM e collaboratore dello studio. “Nei muscoli, questo può significare che le proteine necessarie per la loro formazione, l’integrità e la funzione non si trovano al posto giusto nella cellula”. Nei loro esperimenti in coltura cellulare, i ricercatori hanno potuto riavviare il trasporto con una certa sostanza attiva.Questo potrebbe essere un punto di partenza per lo sviluppo di farmaci per il trattamento di questa malattia ereditaria grave e attualmente incurabile.
Fonte:http://medicalxpress.com/news/2016-01-scientists-unravel-defects-rare-hereditary.html