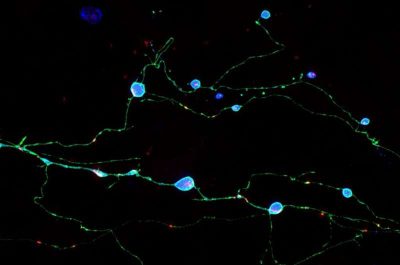
(Malattie da prioni-Immagine:neuroni cresciuti in coltura che esprimono una proteina prionica mutante (ciano) che causa malattie da prioni nell’uomo. Questi neuroni mostrano assoni rigonfi che contengono aggregati di proteine prioniche mutanti tossiche. Chassefeyre et al. hanno identificato i geni che spiegano la formazione di questi aggregati e hanno dimostrato che la riduzione della loro funzione può inibire la formazione degli aggregati e prevenire la disfunzione neuronale. Credito: Adriaan Verhelle e Yin Wu (Ricerca Scripps) ).
Le malattie da prioni, come la malattia di Creutzfeldt-Jakob (CJD), sono sindromi di demenza fatale associate alla formazione di aggregati della proteina prionica, PrP. Il modo in cui questi aggregati si formano e uccidono le cellule cerebrali non è mai stato completamente compreso, ma un nuovo studio degli scienziati di Scripps Research suggerisce che gli aggregati uccidono i neuroni danneggiando i loro assoni, le strette fibre nervose attraverso le quali inviano segnali ad altri neuroni.
L’accumulo di aggregati proteici negli assoni, insieme a rigonfiamenti assonali e altri segni di disfunzione, sono anche caratteristiche precoci di altre malattie neurodegenerative tra cui il morbo di Alzheimer e il morbo di Parkinson. La scoperta di come si formano questi aggregati di prioni negli assoni e di come inibirli, riportata in Science Advances, potrebbe in definitiva avere un significato che va ben oltre le malattie da prioni.
“Speriamo che questi risultati portino a una migliore comprensione dei prioni e di altre malattie neurodegenerative, nonché a nuove strategie per curarle”, afferma l’autore senior dello studio Sandra Encalada, Ph.D., Arlene e Arnold Goldstein Professore Associato nel Dipartimento di Medicina Molecolare presso lo Scripps Research.
I ricercatori nel loro studio hanno osservato da vicino copie mutanti e patogene della proteina prionica PrP che formano grandi aggregati negli assoni dei neuroni, ma non nei principali corpi cellulari dei neuroni. La formazione di questi aggregati è stata seguita da segni di disfunzione assonale e infine morte neuronale. Gli scienziati hanno trovato prove che i processi di smaltimento dei rifiuti dei neuroni normalmente sono in grado di far fronte a tali aggregati quando si trovano all’interno o vicino ai principali corpi cellulari dei neuroni, ma sono molto meno in grado di farlo quando gli aggregati si accumulano lontano all’interno degli assoni.
I ricercatori hanno anche identificato un complesso di proteine chiave responsabile della trasmissione della PrP negli assoni e della causa dell’aggregazione associata a grandi rigonfiamenti assonali. Hanno dimostrato che silenziando una qualsiasi di queste proteine potrebbero inibire la formazione degli aggregati e proteggere i neuroni da danni e morte.
Vedi anche:Test diagnostico rivoluzionario per le malattie da prioni
Assoni vulnerabili
La CJD è la malattia da prioni umana più comune, che si verifica al ritmo di circa un caso per milione di persone all’anno in tutto il mondo. Si pensa che la maggior parte dei casi si presenti spontaneamente quando la PrP in qualche modo viene alterata nel cervello e inizia ad aggregarsi. Poiché questi aggregati crescono attraverso un processo di reazione a catena che attira copie sane di PrP, in rari casi possono trasmettere CJD, ad esempio durante un intervento chirurgico di trapianto di cornea, da una persona all’altra. Circa il 15% dei casi è ereditario, causato da mutazioni che rendono più probabile l’aggregazione della PrP. I disturbi da prioni si verificano in altri mammiferi e si pensa che siano dovuti a simili aggregazioni tossiche di proteine PrP di specie diverse.
Nello studio, il team di Encalada ha utilizzato cellule cerebrali di topo contenenti PrP mutante, insieme a tecniche microscopiche, per studiare l’accumulo iniziale di aggregati di PrP negli assoni. L’assone di un neurone è spesso molto lungo rispetto al suo corpo principale, il soma, ed è stato scoperto che è particolarmente vulnerabile alle interruzioni dei suoi delicati sistemi per il trasporto di molecole essenziali e l’eliminazione dei rifiuti.
La normale funzione della PrP nei neuroni non è mai stata chiara, ma la proteina sembra essere normalmente secreta, attraverso contenitori simili a sacche chiamati vescicole, dal soma e dall’assone, dove a volte ritorna per essere riciclata o degradata come rifiuto. I ricercatori hanno scoperto nei loro esperimenti che la PrP mutante prodotta nel soma è anche in gran parte incapsulata in vescicole che vengono spostate nell’assone lungo binari chiamati microtubuli.
Questo movimento coinvolge un sistema di traffico di vescicole alquanto complesso e i ricercatori hanno osservato che questo sistema devia gran parte della PrP negli assoni, dove le vescicole contenenti PrP si raccolgono e si fondono. La PrP mutante in questa situazione forma grandi aggregati – Encalada li chiama endoggresomi – di cui gli assoni non possono liberarsi. Gli aggregati portano a rigonfiamenti assonali e altri segni di disfunzione tra cui una ridotta segnalazione del calcio neuronale e, in definitiva, un tasso di morte neuronale molto più rapido rispetto ai neuroni con PrP normale.
I ricercatori hanno anche trovato un modo per contrastare la formazione di endoggresomi. Hanno identificato quattro proteine, Arl8, kinesin-1, Vps41 e SKIP, che sono responsabili della direzione delle vescicole contenenti PrP negli assoni, portandole lontano nel soma e fondendole con altre vescicole contenenti PrP per innescare la formazione di aggregati. Quando hanno messo a tacere una di queste proteine, molte meno vescicole contenenti PrP sono entrate negli assoni, gli assoni hanno mostrato pochi o nessun segno di aggregazione e i neuroni hanno funzionato normalmente o quasi normalmente e sono sopravvissuti proprio come le normali cellule cerebrali.
I risultati indicano la possibilità allettante che le malattie da prioni, e forse molte altre malattie del cervello da aggregati proteici, possano essere prevenute o trattate interrompendo almeno transitoriamente il processo di traffico che porta le proteine incapsulate nelle vescicole e inclini agli aggregati negli assoni.
“Siamo molto entusiasti di scoprire molecole in grado di inibire questo percorso di formazione di aggregati e di studiare gli effetti di tali inibitori in modelli animali di prioni e altre malattie neurodegenerative“, afferma Encalada.
“Endosomal Sorting Drives the Formation of Axonal Prion Protein Endogresomes” è stato co-autore di Romain Chassefeyre, Tai Chaiamarit, Adriaan Verhelle, André Leitão e Sandra Encalada, tutti di Scripps Research; e Sammy Weiser Novak, Leonardo Andrade e Uri Manor, del Salk Institute for Biological Studies.
Fonte:Science