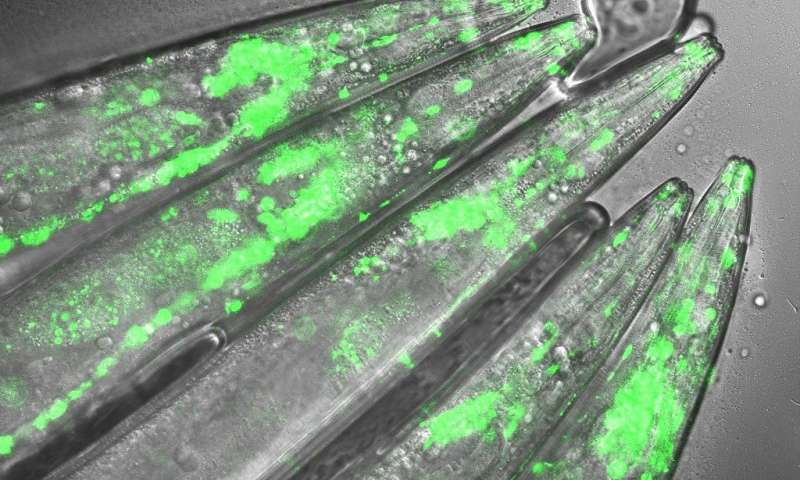
Immagine: un modello di C. elegans della malattia di Huntington sotto il microscopio. Quando gli scienziati mettono a tacere la proteina UBR5, vedono un drammatico aumento nel numero di aggregati di proteine tossiche nei neuroni. Le aree verdi sono gli aggregati proteici. Credito: Seda Koyuncu e Isabel Saez.
Il neuroscienziato Dr. David Vilchez e il suo team al CECAD, il Cluster of Excellence for Aging Research dell’Università di Colonia, hanno compiuto un passo importante verso la comprensione dei meccanismi che causano il disturbo neurodegenerativo della malattia di Huntington. Nello specifico, hanno identificato un sistema che blocca l’accumulo di aggregati di proteine della tossina, responsabili della neurodegenerazione.
I risultati sono stati pubblicati sulla rivista Nature Communications.
La malattia di Huntington è una malattia neurodegenerativa che provoca la morte delle cellule cerebrali, portando a movimenti incontrollati del corpo, perdita della parola e psicosi. Le mutazioni nel gene huntingtina causano la malattia, determinando l‘aggregazione tossica della proteina huntingtina. L’accumulo di questi aggregati provoca la neurodegenerazione e solitamente porta alla morte del paziente entro 20 anni dall’esordio della malattia.
Per esaminare i meccanismi alla base della malattia di Huntington, Vilchez e il suo gruppo hanno usato le cosiddette cellule staminali pluripotenti indotte (iPSC) dai pazienti con malattia di Huntington, che sono in grado di differenziarsi in qualsiasi tipo di cellula, come i neuroni. Le cellule staminali pluripotenti indotte derivate da pazienti con malattia di Huntington mostrano una sorprendente capacità di evitare l’accumulo di aggregati di proteine tossiche, un segno distintivo della malattia. Anche se le iPSC esprimono il gene mutante responsabile della malattia di Huntington, non sono stati trovati aggregati.
I ricercatori hanno identificato una proteina chiamata UBR5 come meccanismo protettivo delle cellule, promuovendo la degradazione dell’huntingtina mutata. Questi risultati possono contribuire a una migliore comprensione della malattia di Huntington e potrebbero essere un trampolino di lancio per lo sviluppo di ulteriori trattamenti nei pazienti.
( Vedi anche:Trovato nuovo pezzo importante nel puzzle della malattia di Huntington).
I ricercatori hanno esaminato le iPSC da pazienti e neuroni derivati, per differenze nella loro capacità di evitare l’aggregazione huntingtina mutante. Hanno scoperto che l’huntingtina può essere degradata dal sistema di smaltimento cellulare noto come proteasoma. Tuttavia, questo sistema è difettoso nei neuroni, il che porta alla aberrante aggregazione della proteina huntingtina mutata. Vilchez e il suo team hanno scoperto che UBR5 è aumentata nelle cellule staminali pluripotenti per accelerare la degradazione dell’ huntingtina nelle cellule. Per esaminare il ruolo di UBR5 nella regolazione del gene mutante huntingtina (HTT), i ricercatori hanno ridotto i livelli di UBR5 e hanno potuto immediatamente vedere un accumulo di proteine aggregate nelle iPSC.
“È stato sorprendente”, afferma Vilchez. “Dal nulla, le celle hanno raggiunto enormi quantità di aggregati”.
Gli autori hanno compiuto un ulteriore passo avanti e hanno esaminato se UBR5 controlla anche l’aggregazione huntingtina mutante nei modelli di malattia di Huntington. Hanno scoperto che la disregolazione di UBR5 determina un massiccio aumento dell’aggregazione e degli effetti neurotossici nei neuroni. D’altra parte, promuovere l’attività UBR5 blocca l’ aggregazione huntingtina mutante nei modelli di malattia di Huntington.
Per testare la specificità dei risultati, i ricercatori hanno tenuto d’occhio anche altre malattie. “Abbiamo anche verificato il meccanismo in altre malattie neurodegenerative come la sclerosi laterale amiotrofica “, afferma Seda Koyuncu, un dottorando che lavora nel laboratorio di Vilchez e autore principale della pubblicazione. “Il nostro risultato è molto specifico per la malattia di Huntington“, aggiunge la dottoressa Isabel Saez, un altro autore principale che lavora con Vilchez al CECAD.
Fonte: Nature