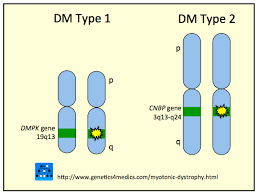
“La distrofia miotonica (DM) è una malattia genetica neuromuscolare degenerativa a carattere autosomico dominante, caratterizzata da un quadro clinico ampiamente variabile e da un decorso lentamente progressivo, il cui esordio può avvenire a qualunque età. Rappresenta la seconda forma di distrofia muscolare più diffusa dopo la distrofia muscolare di Duchenne.
Il quadro clinico è caratterizzato da perdita di massa muscolare, miotono, cataratta, difetti nel sistema di conduzione cardiaco, alterazioni endocrine e deficit cognitivi nei casi congeniti. È presente un fenomeno di anticipazione per cui l’esordio tende a presentarsi ad un’età sempre più giovane di generazione in generazione nella stessa famiglia“.
I sintomi di questa condizione sono legati all’accumulo di trascritti genici tossici nelle cellule muscolari che derivano da anomalie dello splicing che in biologia molecolare e in genetica, (dall’inglese: montaggio) è una modifica del nascente pre-mRNA che avviene insieme o dopo la trascrizione, nella quale gli introni, ossia le regioni non codificanti del gene, vengono rimossi e gli esoni che sono la porzione di un gene che viene trascritta dall’ RNA polimerasi durante il processo di trascrizione, vengono uniti. Ciò è necessario per il tipico RNA messaggero prima che possa essere usato per produrre una corretta proteina tramite la traduzione o sintesi proteica..
( Vedi anche: Importante passo avanti nella comprensione della distrofia muscolare).
Recenti studi hanno indicato che la salute delle cellule muscolari e la funzione dipendono in modo critico dalle vie che supportano il bilancio energetico e l’autofagia, un processo che aiuta a degradare e riciclare i detriti cellulari.
Uno studio condotto da Perrine Castets presso l’Università di Basilea ha dimostrato che i trattamenti farmacologici che hanno come obiettivo AMPK e vie di segnalazione mTOR, che regolano il bilancio energetico e l’autofagia nelle cellule, hanno migliorato i sintomi della distrofia miotonica in un modello murino. Inizialmente i ricercatori hanno osservato che i percorsi AMPK e mTORC1 risultano interrotti nel tessuto muscolare dei topi modello di MD1.
Ulteriori indagini hanno rivelato che anche l’autofagia è stata compromessa nel muscolo MD1 e questo ha contribuito allo sviluppo dei sintomi distrofia-simili nei topi. Quando i topi MD1 sono stati trattati con un farmaco che attiva la segnalazione AMPK, essi hanno mostrato miglioramenti nella funzione muscolare così come riduzioni di anormale splicing del gene.
Trattare i topi MD1 con la Rapamicina, un farmaco clinicamente approvato che attiva la segnalazione mTORC1, ha anche ridotto i segni di patologia muscolare. Questi risultati identificano obiettivi nei percorsi AMPK e mTORC1 che possono essere utilizzati per potenziali terapie per il trattamento della distrofia miotonica.
Fonte: EurekAlert