Per la prima volta, i ricercatori dell’ Università di Pittsburgh School of Medicine hannp identificato due geni responsabili della sindrome del cuore sinistro ipoplasico (HLHS).
I risultati dello studio sono stati pubblicati oggi sulla rivista Nature Genetics.
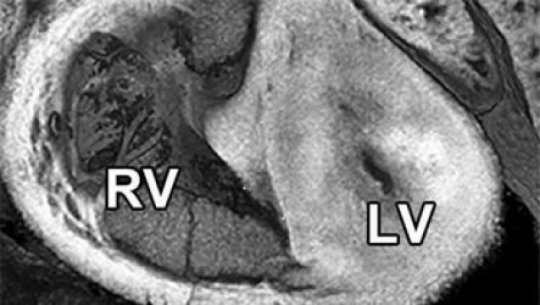
Le malattie cardiache congenite o anomalie strutturali del cuore che sono presenti alla nascita, riguardano fino all’ 1 per cento di tutti i nati vivi. La sindrome del cuore sinistro ipoplasico è una cardiopatia congenita rara in cui il lato sinistro del cuore è poco sviluppato, con conseguente incapacità di pompare il sangue in modo efficace al resto del corpo. Essa si verifica in circa il 2-3 nati vivi su 10.000 negli Stati Uniti ed è fatale se non trattata.
L’attuale trattamento della HLHS coinsiste in molteplici interventi chirurgici complessi nei primi anni di vita di un bambino e mentre è efficace in molti pazienti, non migliora la funzione cardiaca in molti altri causando insufficienza cardiaca e la necessità di trapianto di cuore. Il tasso di sopravvivenza a cinque anni per i pazienti con sindrome del cuore sinistro ipoplasico è circa del 50/70 per cento.
Anche se i fattori di rischio genetici sono noti per svolgere un ruolo nella HLHS, geni specifici sono stati difficili da identificare.
“Studiare le malattie con una genetica complessa è estremamente impegnativo. Il nostro studio è stato reso possibile sfruttando risultati di un’analisi su larga scala condotta sui topi per individuare le mutazioni che causano difetti cardiaci congeniti. Ciò ha comportato il recupero dei primi modelli murini di HLHS. L’ analisi di questi topi con HLHS ha permesso di individuare per la prima volta, due geni che interagiscono in combinazione per causare la sindrome del cuore sinistrio ipoplasico “, ha detto Cecilia lo, Prof. alla Scuola di Medicina della Pitt University che dirige anche la F. Sargent Cheever in Developmental Biology. ” La comprensione della genetica e della biologia di questa condizione questo può facilitare lo sviluppo di nuove terapie per migliorare la prognosi dei pazienti”.
Io ed il suo team hanno usato l’ ecografia del feto per lo screening di topi con mutazioni indotte sperimentalmente, alla ricerca di difetti cardiaci strutturali. Da questa schermata, hanno recuperato otto diversi ceppi di topi con difetti cardiaci strutturali e funzionali indicativi della HLHS.
Confrontando il genoma dei topi con difetti cardiaci HLHS con il genoma dei topi normali, i ricercatori hanno identificato diverse centinaia di mutazioni nei ceppi mutanti HLHS. Ulteriori analisi di questi topi mutanti hanno indicato le origini genetiche della HLHS che probabilmente coinvolge molti geni interagenti. La rilevanza di queste mutazioni era rafforzata dal fatto che molte mutazioni sono state trovate anche nelle stesse regioni cromosomiche che erano state associate alla sindrome da studi genetici umani.
In un ceppo di topi, i ricercatori hanno scoperto che le mutazioni in due geni, chiamati Sap130 e Pcdha9, erano necessarie per lo sviluppo della HLHS.
“È interessante notare, che la HLHS è stata trovata solo in animali con mutazioni in entrambi i geni. Tuttavia, gli animali che avevano mutazioni in Pcdha9, ma non in Sap130, mostravano difetti nell’aorta, ma ventricolo sinistro di dimensioni normali, suggerendo che le interazione tra i due geni erano necessarie per provocare tutte le caratteristiche della HLHS “, ha detto Xiaoqin Liu, primo autore del nuovo studio e istruttore di ricerca nel laboratorio di lo.
Utilizzando l’editing genetico CRISPR-Cas9 nei topi, i ricercatori hanno confermato che le mutazioni in questi due geni possono causare la sindrome del cuore sinistro ipoplasico. Inoltre, hanno sequenziato 68 campioni di HLHS da pazienti e trovato un individuo con rare mutazioni in entrambi i geni SAP130 e PCDHA.
L’analisi molecolare ha dimostrato che le cellule del cuore negli animali colpiti dalla malattia, sono poco sviluppate e avevano difetti mitocondriali, indicando che le interazioni Sap130-Pcdha9 svolgono un ruolo cruciale non solo per lo sviluppo del cuore, ma anche nella regolazione della funzione metabolica del muscolo cardiaco.
Questi risultati suggeriscono che la sindrome può essere associata ad un difetto cellulare fondamentale nel muscolo cardiaco che può compromettere il flusso di sangue nei pazienti. “La scoperta ha importanti implicazioni terapeutiche in quanto la riparazione chirurgica del cuore non sarà in grado di affrontare e risolvere il difetto muscolare cellulare nei pazienti HLHS”, ha osservato Io.
I ricercatori stanno studiando altre mutazioni identificate attraverso lo screening genetico e intraprendendo ulteriori analisi genetiche dei pazienti HLHS per comprendere meglio i fattori molecolari e genetici che causano la malattia.
Fonte: Nature