La malattia di Huntington (HD) è una malattia neurodegenerativa a trasmissione dominante caratterizzata dall’espansione del numero di ripetizioni CAG trinucleotidiche nel primo esone del gene HTT che codifica per la proteina huntingtina. Mentre il numero normale di queste ripetizioni varia tra 6 e 35, la patologia si osserva quando il numero raggiunge un massimo di circa 150 ripetizioni.
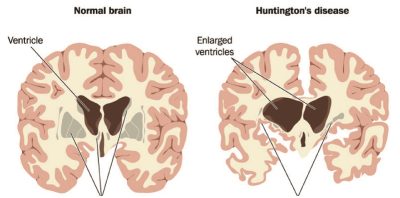
La manifestazione anatomica più ovvia della malattia è una perdita selettiva di neuroni mediamente spinosi nello striato, che presumibilmente porta a un numero qualsiasi di anomalie motorie. Mentre i ricercatori stanno finalmente iniziando a comprendere i meccanismi con cui le ripetizioni vengono espanse e portano a danni, altri risultati accidentali legati all’Huntington, come l’attivazione degli elementi trasponibili, sono probabilmente responsabili di gran parte della variabilità aggiuntiva osservata indipendentemente dalla lunghezza della ripetizione.
Normalmente, si potrebbe associare l’attivazione del retrotrasposone con un esito casuale e di conseguenza indesiderabile, come potrebbe verificarsi nei tumori o in una serie di altre malattie neurodegenerative. Tuttavia, ci sono anche molti effetti utili e talvolta essenziali dell’attivazione degli elementi trasponibili nel cervello, sia durante la neurogenesi (come nella diversificazione somatica e il successivo mosaicismo neurale), sia nei processi in corso come la riparazione del DNA, il sonno e la formazione della memoria.
Vedi anche:SLA e Malattia di Huntington: CRISPR-Cas13 prende di mira le proteine mutanti
In un recente articolo apparso su Frontiers in Cellular Neuroscience, i ricercatori hanno intrapreso un’analisi bioinformatica dei dati pubblici di RNA-seq di un pannello di modelli murini HD e sono stati in grado di dimostrare che una diminuzione dell’espressione dell’RNA del retrotrassposone della linea 1 (L1) è associata a due segni distintivi della malattia. Vale a dire, è correlata alla lunghezza della ripetizione CAG e si verifica specificamente nello striato, il sito della neurodegenerazione. Questi risultati sono stati poi convalidati sperimentalmente in topi knock-in Htt e in cervelli umani post mortem. Questi risultati sembrerebbero implicare che potrebbe essere necessario un certo livello di L1 per evitare l’Huntington nelle persone che potrebbero essere geneticamente suscettibili.
Le espansioni ripetute di triplette CAG sono generalmente causate dallo slittamento durante la replicazione del DNA. Ciò porta alla formazione di strutture “loop out” al fine di mantenere l’accoppiamento di basi complementari tra il filo genitore e il filo figlia che viene sintetizzato. Se la struttura del loop out è formata dalla sequenza sul filo figlia, ciò comporterà un aumento del numero di ripetizioni. Al contrario, se la struttura loop out si forma sul filo genitore, si verifica una diminuzione del numero di ripetizioni. Le cose si fanno interessanti quando il macchinario di riparazione del DNA viene coinvolto per cercare di riparare eventuali regioni danneggiate.
A seconda della natura del danno e degli strumenti a disposizione della cellula in quel momento, possono essere implementati processi di ricombinazione omologa, giunzione di estremità non omologhe, riparazione del disadattamento o riparazione dell’escissione della base. In tutti questi eventi è richiesta una fase di sintesi del DNA, accompagnata dalla possibilità di slittamento del filamento, che porta a un’ulteriore espansione della ripetizione del trinucleotide. Ci sono alcune prove che lo slittamento può essere osservato somaticamentenel cervello, al contrario del solo DNA delle cellule germinali ereditabili, nel qual caso il cervello diventerebbe un mosaico. L’osservazione che i retrotrasposoni e gli elementi retrovirali endogeni sono intimamente legati ai processi di riparazione e sono attivati in modo simile a mosaico durante il corso dello sviluppo cerebrale porta alla domanda: c’è un ruolo significativo per questi elementi direttamente nell’espansione ripetuta?
Questa potrebbe essere una domanda per un’analisi successiva; tuttavia, ti lasceremo con un’ulteriore nuova allettante scoperta. L’ultima ricerca suggerisce che l’origine della mielina stessa potrebbe essere un risultato diretto delle attività di un retrotrasposone noto come RNLTR12-int. L’RNA non codificante di questo elemento si lega al fattore di trascrizione SOX10, che regola in modo critico la trascrizione della proteina di base della mielina, che a sua volta è l’invenzione proteica che definisce la mielinizzazione a spirale avanzata in stile vertebrato.