Huntington-Immagine:ìCredit EMBO Molecular Medicine.
Gli scienziati del Weizmann Institute hanno scoperto due piccole molecole che possono attraversare la barriera ematoencefalica nei topi, rallentando e persino invertendo gli effetti della malattia di Huntington, che è incurabile.
Il cervello umano è un centro di controllo ben custodito. Il suo sistema di vasi sanguigni è circondato da una barriera cellulare densamente compattata che impedisce alla maggior parte delle sostanze di entrare o uscire. Questa architettura fortificata protegge il cervello, ma può anche impedirgli di ricevere aiuto quando ne ha bisogno, ad esempio nel caso di una malattia neurodegenerativa.
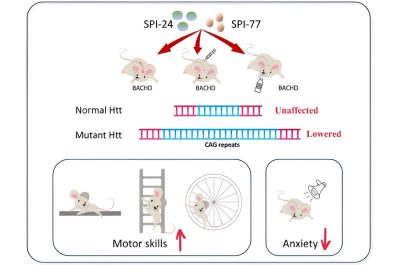
In un nuovo studio, pubblicato su EMBO Molecular Medicine, la Prof. Rivka Dikstein del Weizmann Institute of Science e il suo team hanno identificato due piccole molecole che riescono a penetrare la barriera emato-encefalica e a ridurre i livelli di una proteina difettosa che causa la malattia di Huntington, una malattia neurodegenerativa incurabile. I nuovi farmaci non solo hanno rallentato la progressione della malattia nei modelli murini, ma hanno anche invertito alcuni dei suoi sintomi.
I segni e i sintomi della malattia di Huntington (lievi movimenti involontari, goffaggine generale e aumento dell’ansia) emergono più comunemente intorno ai 40 anni. La malattia si sviluppa nel tempo e porta inevitabilmente alla morte. È causata da un’eccessiva ripetizione, 36 volte o più, di un segmento di DNA nel gene dell’huntingtina.
Le persone con la malattia di Huntington di solito hanno una copia funzionante del gene dell’huntingtina e una copia difettosa, che porta alla creazione di proteine difettose che si attaccano insieme, formando un residuo tossico nel cervello. Questo residuo si accumula e danneggia il cervello in vari modi, provocando infiammazioni, ostacolando l’espressione di geni vitali per la sopravvivenza delle cellule nervose e danneggiando le centrali elettriche cellulari chiamate mitocondri.
Gli sforzi precedenti per trattare ciascun meccanismo separatamente non erano sufficientemente efficaci, mentre i farmaci volti ad affrontare la radice del problema – la stessa proteina Huntingtina difettosa – trovavano difficile distinguere la proteina difettosa da quella normale.
Un possibile raggio di speranza è stato scoperto nel 2019 dalla Dottoressa Anat Bahat, ricercatrice nel laboratorio di Dikstein nel dipartimento di scienze biomolecolari di Weizmann. Da anni il laboratorio conduce ricerche di base sulla proteina regolatrice Spt5, una grande proteina con molti segmenti funzionali che aiuta nella produzione di molecole di RNA messaggero ed è particolarmente importante nella produzione di proteine coinvolte nell’infiammazione.
Bahat e colleghi hanno scoperto che diverse piccole molecole possono inibire alcune funzioni di Spt5 senza causare danni significativi alle altre funzioni della proteina. I ricercatori hanno poi scoperto tre piccole molecole che inibivano specificamente l’espressione dell’huntingtina mutata senza danneggiare l’espressione dell’huntingtina normale e di altre proteine legate all’infiammazione.
Nel nuovo studio, Bahat ha guidato un team di ricercatori che ha esaminato 17 molecole con strutture chimiche simili a quelle trovate efficaci nello studio precedente, nella speranza di identificare gli inibitori più efficaci dell’espressione dell’huntingtina mutata. Utilizzando colture cellulari di un modello murino della malattia di Huntington, i ricercatori hanno identificato le due molecole più efficaci.
Hanno poi testato le nuove molecole in colture cellulari di pazienti umani con vari gradi di difetto genetico e, in ogni caso, hanno identificato un calo significativo nella quantità di proteina mutante. Allo stesso tempo, non si è verificata alcuna diminuzione nell’espressione della proteina non mutante, prova che le nuove molecole stavano svolgendo il loro lavoro con grande precisione.
La fase successiva dello studio prevedeva di testare le due molecole selezionate in topi geneticamente modificati che esprimevano una versione completamente umana del gene mutante dell’huntingtina. I topi sono stati inizialmente trattati in età avanzata, quando già mostravano chiari segni della malattia, mediante un’infusione somministrata direttamente nella parte danneggiata del cervello nel corso di quattro settimane. Il trattamento ha ridotto l’espressione della copia mutante del gene e ha aumentato la percentuale delle proteine sane interessate nell’area danneggiata del cervello.
Il trattamento è riuscito anche ad alleviare alcuni dei danni causati dalla malattia. Ha aumentato l’espressione di due geni che di solito vengono danneggiati man mano che la malattia progredisce: uno è un fattore di crescita essenziale per la sopravvivenza delle cellule nervose e l’altro appartiene ai mitocondri.
“Anche se il modello murino invecchiato simulava la malattia in uno stadio avanzato“, spiega Bahat, “il trattamento è riuscito in una certa misura a riportare indietro le lancette del tempo. Negli studi comportamentali abbiamo osservato livelli ridotti di ansia e miglioramento dell’equilibrio e della coordinazione nei topi trattati“.
Tuttavia, la somministrazione di un’infusione direttamente nell’area danneggiata del cervello richiede una procedura chirurgica complessa, rischiosa e dolorosa per i pazienti. I ricercatori hanno quindi cercato di stabilire se il farmaco potesse essere efficace se somministrato sotto forma di pillola o tramite iniezione sottocutanea.
Sono stati entusiasti di scoprire che questi metodi alternativi erano riusciti anche a ridurre i livelli di Huntingtina mutata nel cervello dei topi e che le molecole riuscivano a completare il lungo viaggio dalla bocca o dalla pelle al cervello senza subire alcun cambiamento significativo. Le loro caratteristiche permettono loro di attraversare la barriera ematoencefalica ed entrare direttamente nella zona malata una volta arrivati a destinazione.
Una di queste piccole molecole ha dimostrato un effetto curativo a dosi particolarmente basse, una proprietà importante per un farmaco progettato per uso umano. Il suo svantaggio, però, era che aveva effetto su più di 1.000 altri geni. La seconda molecola richiedeva dosi più elevate, ma si è rivelata un’arma relativamente mirata: ha ridotto l’espressione del gene mutante senza causare effetti collaterali o modifiche diffuse ad altri geni.
Nella parte finale dello studio i ricercatori hanno esaminato gli effetti del trattamento somministrato per via orale nel corso di due mesi, nella fase iniziale, quando erano appena comparsi i primi segni della malattia. Tutti i topi malati mostravano livelli elevati di ansia prima dell’inizio dell’esperimento, ma in quelli che hanno ricevuto il trattamento, l’ansia è tornata a livelli normali.
Nel corso del tempo, la perdita di equilibrio e l’iperattività dei topi malati nel gruppo di controllo sono peggiorate, mentre quelli trattati hanno riportato danni meno sostanziali all’equilibrio e non hanno manifestato iperattività. Questi risultati, insieme ad altri test, hanno indicato che i nuovi farmaci ritardano il progresso della malattia, anche se somministrati in una fase precoce.
“Siamo stati lieti di scoprire che piccole molecole riuscivano a raggiungere il cervello senza subire cambiamenti e senza disintegrarsi lungo il percorso“, afferma Dikstein. “Mentre altri trattamenti sperimentali richiedono ripetuti interventi chirurgici al cervello o alla colonna vertebrale, queste molecole, somministrate per via orale o mediante iniezione, potrebbero aprire la strada a un trattamento efficace e sicuro della malattia di Huntington”.
Leggi anche:Huntington: dieta ricca di fibre ritarda i sintomi
“Negli ultimi anni è diventato chiaro che una certa funzione di grandi proteine regolatrici può essere mirata con precisione utilizzando minuscole molecole, senza danneggiare il funzionamento complessivo di queste proteine. Questa comprensione potrebbe gettare le basi per nuovi trattamenti per una varietà di malattie“.
Fonte:EMBO Molecular Medicine