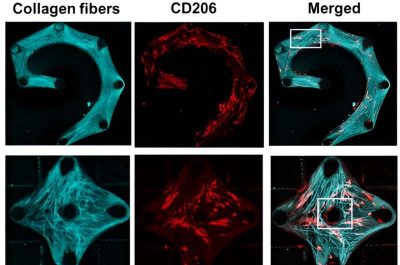
Fibrosi polmonare-Le immagini mostrano fibre di collagene (colonna di sinistra) e proteine della superficie cellulare che agiscono come marcatori per i macrofagi (colonna centrale). La colonna di destra mostra i macrofagi allineati con le fibre di collagene. Credito: Università di Buffalo.
La fibrosi polmonare, come si osserva nella fibrosi polmonare idiopatica (IPF) e nella fibrosi polmonare indotta da COVID, è una malattia polmonare grave e spesso fatale ed é caratterizzata da cambiamenti sostanziali nell’architettura, nella composizione e nella rigidità del tessuto polmonare che portano al deterioramento delle funzioni polmonari. Inizia a seguito di una lesione epiteliale alveolare, seguita dal reclutamento e dall’attivazione di cellule immunitarie e dalla conseguente guarigione aberrante della ferita.
Nei polmoni fibrotici è stato segnalato un aumento dell’accumulo di macrofagi, comprese sottopopolazioni pro-fibrotiche uniche. Tuttavia, il contributo dei diversi stati di attivazione dei macrofagi alla progressione e alla gravità della malattia non è ancora chiaro. Ad esempio, i macrofagi possono contribuire alla normale riparazione o ottenere ruoli pro-fibrotici, a seconda dei tempi, del contesto e del loro stato di attivazione. È stato dimostrato che l’assenza di macrofagi durante l’infiammazione determina una compromissione della guarigione delle ferite, mentre la persistenza dei macrofagi oltre le fasi di riparazione acuta può provocare fibrosi degli organi.
Studi recenti suggeriscono che quest’ultimo effetto è dovuto almeno in parte al contributo dei macrofagi pro-fibrotici all’attivazione dei fibroblasti nei miofibroblasti in un processo che dipende dal contatto fisico. I miofibroblasti sono le principali cellule effettrici della fibrogenesi poiché producono quantità eccessive di collagene e generano elevate forze contrattili incorporando l’actina del muscolo liscio α (α-SMA) nelle fibre da stress. Tuttavia, non è ancora chiaro come la comunicazione macrofago-miofibroblasto a livello tissutale contribuisca alla fibrogenesi del tessuto polmonare.
Gli scienziati sanno da tempo che i globuli bianchi chiamati macrofagi si accumulano nei polmoni delle persone affette da fibrosi polmonare. Il ruolo svolto dai macrofagi nello sviluppo della malattia polmonare, spesso fatale, è meno chiaro.
Un nuovo studio condotto dall’Università di Buffalo fa luce su questo mistero e apre nuove strade per studiare la fibrosi polmonare, progressi che potrebbero alla fine portare a medicine e terapie più efficaci per la malattia, che colpisce circa 100.000 persone negli Stati Uniti.
“La nostra comprensione di come si sviluppa la fibrosi polmonare è notevolmente migliorata; tuttavia, c’è ancora molto che non capiamo, in particolare il coinvolgimento delle cellule immunitarie nello sviluppo della malattia”, afferma l’autore corrispondente dello studio Ruogang Zhao, Ph.D., Professore associato di ingegneria biomedica presso l’Università di Buffalo.
Nelle persone affette da fibrosi polmonare, nei polmoni si forma tessuto cicatriziale rigido che rende difficile la respirazione. Questo tessuto cicatrizzato rigido non può essere riparato, ma solo rallentato con farmaci e terapie.
Lo studio, pubblicato su Science Advances, descrive come Zhao e colleghi hanno sviluppato modelli in miniatura di tessuti polmonari fibrotici che fungono da proxy per qualcuno con fibrosi polmonare.
Oltre ai macrofagi, il modello includeva i due componenti principali del tessuto cicatriziale rigido: fibroblasti e fibre di collagene.
Durante gli esperimenti, il team ha osservato che i macrofagi percepivano l’ambiente circostante e si “allineavano” con i fibroblasti e le fibre di collagene. Questa attivazione della sensibilità meccanica dei macrofagi ha permesso loro di secernere ulteriori fattori biochimici che promuovono la crescita del tessuto cicatriziale.
Il team ha poi somministrato al tessuto malato un farmaco chiamato Pirfenidone, un trattamento approvato dalla Food and Drug Administration che rallenta il peggioramento della fibrosi polmonare. Il Pirfenidone blocca alcune proteine che possono influenzare in modo significativo il modo in cui i macrofagi aderiscono e interagiscono con i fibroblasti e le fibre di collagene. Di conseguenza, il farmaco può impedire la formazione di tessuto cicatriziale.
“I risultati suggeriscono un potenziale meccanismo, a livello tissutale e che coinvolge i macrofagi, su come si origina la fibrosi polmonare”, afferma il primo autore dello studio Ying Xu, Ph.D. candidato nel laboratorio di Zhao.
Leggi anche:Fibrosi polmonare: inibitori DPP4 p rigenerano il tessuto polmonare
“Il modello di tessuto polmonare fibrotico”, aggiunge Zhao, “è un nuovo potente strumento che i ricercatori possono utilizzare per studiare ulteriormente le interazioni a livello di tessuto e cellula nei polmoni fibrotici. Potrebbe anche aiutare a testare nuovi farmaci che rallentano ulteriormente la malattia o la fermano. Insieme, queste analisi suggeriscono che i macrofagi polmonari subiscono un’attivazione meccanica all’interno del microambiente di tessuto fibrotico ampiamente rimodellato dei polmoni IPF umani”.
Fonte:Science Advances