(Diabete-Immagine:le cellule beta (verdi) producono l’ormone insulina. Livelli elevati di micro RNA 200 sono dannosi. Credito: Masur/Wikimedia Commons).
Una caratteristica centrale del diabete di tipo 1 è la perdita delle cellule beta pancreatiche che producono insulina. I ricercatori guidati da Paolo Fiorina, MD, Ph.D., del Boston Children’s Hospital e Francesca D’Addio, MD, dell’Università di Milano, hanno ora identificato un percorso cellulare dannoso che causa la morte di queste cellule. Quando hanno bloccato il percorso nei topi e nelle isole pancreatiche umane, dove risiedono le cellule beta, hanno preservato le cellule beta, aumentato la produzione di insulina e prevenuto o ritardato l’insorgenza del diabete.
Lo studio, pubblicato il 3 febbraio su Nature Communications, ha utilizzato tre diversi modelli murini di diabete e ha riscontrato effetti protettivi indipendentemente dal fatto che il percorso fosse bloccato geneticamente o da un anticorpo. Gli studi su cellule umane e persone con diabete erano coerenti con i risultati ottenuti da studi sui topi. I ricercatori sperano di sviluppare una terapia per bloccare il percorso per il trattamento del diabete di tipo 1.
Il percorso consiste in un “recettore della morte” sulle cellule beta produttrici di insulina chiamato TMEM219, accoppiato con la proteina legante il fattore di crescita insulino-simile 3 (IGFBP3), che interagisce con questo recettore. Quando IGFBP3 si lega a TMEM219, i ricercatori hanno scoperto che le cellule beta muoiono attraverso il processo di apoptosi.
“Riteniamo che questo potrebbe essere un meccanismo naturale per tenere sotto controllo la popolazione di cellule beta“, afferma Fiorina. “Pensiamo che nella malattia, la produzione di IGFBP3 possa essere aumentata, quindi c’è una perdita di cellule beta“.
A sostegno di questa idea, il team ha studiato diversi gruppi di persone con diabete e ha riscontrato livelli aumentati di IGFBP3 che circolano nel sangue rispetto ai non diabetici. I livelli sono risultati aumentati anche nelle persone a rischio di sviluppare il diabete e nei topi diabetici e prediabetici.
Vedi anche:Diabete tipo 2: nuovo approccio promettente per la prevenzione
Mantenere in vita le cellule beta
Nei topi, i ricercatori hanno utilizzato diversi mezzi per bloccare il percorso IGFBP3/TMEM219: bloccare IGFBP3, eliminare il gene del recettore TMEM219 o utilizzare una proteina ricombinante basata su parte di TMEM219. Ciascun approccio ha preservato le cellule beta, aumentato la produzione di insulina e ritardato o prevenuto il diabete. Quando il team ha bloccato il percorso per un periodo di tempo più lungo, le popolazioni di cellule beta si sono espanse.
Fiorina e colleghi hanno anche studiato le isole pancreatiche umane, in cui risiedono le cellule beta. Quando hanno esposto le cellule all’IGFBP3, hanno avuto un più alto tasso di morte per apoptosi. Quando hanno bloccato il percorso IGFBP3/TMEM219, le cellule beta sono state protette e hanno continuato a produrre insulina.
“Il pensiero comune per il diabete di tipo 1 è che sia autoimmune”, afferma Fiorina. “Ma l’immunoterapia non cura completamente il diabete. Pensiamo che anche la disregolazione dell’omeostasi delle cellule beta abbia un ruolo e che l’IGFBP3 agisca come una ‘betatossina‘”.
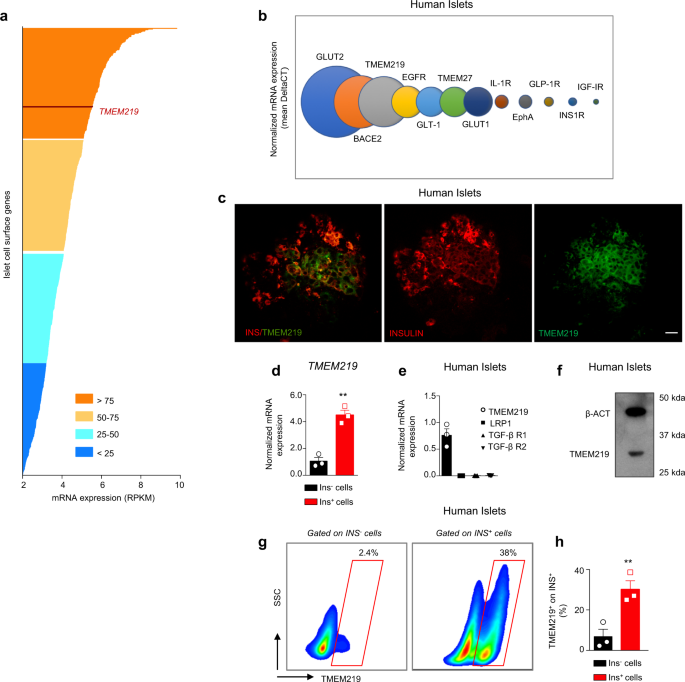
Immagine:a Il profilo del trascrittoma dei geni che codificano per le proteine della superficie delle isole è stato proiettato utilizzando l’RNA-seq di isole isolate da donatori non diabetici ( n = 4). Un elenco completo dei geni analizzati è riportato in Dati supplementari. b Quantificazione dell’espressione relativa di TMEM219 rispetto ad altri recettori rilevanti nelle isole umane mediante qRT-PCR mirata ( n = 4). c Immagine rappresentativa della coespressione TMEM219 (verde) e INS (rosso) in isole umane purificate di donatori non diabetici. Ingrandimento originale ×40, barra della scala 25 μm. Unisci è nel pannello di sinistra. DGrafico a barre che rappresenta l’espressione dell’mRNA di TMEM219 su cellule insulina-positive e negative ordinate per flusso ottenute da isole umane purificate isolate da donatori non diabetici ( n = 3). e Grafici a barre che confrontano l’espressione di TMEM219 e altri recettori putativi IGFBP3 ( LRP1 , TGF-β R1 e TGF-β R2 ) analizzati mediante qRT-PCR in isole umane purificate ( n = 3). f Immunoblot rappresentativo dell’espressione della proteina TMEM219 nelle isole umane (con β-actina come controllo). g , hDiagramma di flusso rappresentativo e un grafico a barre quantitativo che mostra l’espressione di TMEM219 in cellule positive e negative per l’insulina rilevate mediante citometria a flusso in isole umane sane ( n = 3). I dati sono espressi come media ± errore standard della media (SEM) salvo diversa indicazione. ** P < 0,01 dal test t a due code. L’espressione dell’mRNA è stata normalizzata in β-actina ( ACTB ). Gli esperimenti sono stati eseguiti in duplice copia. I dati mostrati in ( c ) e in ( f ) sono il risultato rappresentativo di tre esperimenti indipendenti.
Spiegano gli autori:
“La perdita di cellule beta pancreatiche è una caratteristica centrale del diabete di tipo 1 (T1D) e di tipo 2 (T2D), ma resta da stabilire una strategia terapeutica per preservare la massa delle cellule beta. Qui vi mostriamo che il recettore della morte TMEM219 è espresso sulle cellule beta del pancreas e che la segnalazione attraverso la sua proteina legante il fattore di crescita simile all’insulina ligando 3 (IGFBP3) porta alla perdita e alla disfunzione delle cellule beta. L’aumento dell’IGFBP3 periferico è stato osservato in pazienti con T1D/T2D stabiliti e a rischio ed è stato confermato nei modelli preclinici T1D/T2D, suggerendo che la segnalazione disfunzionale di IGFBP3/TMEM219 è associata ad anomalie nell’omeostasi delle cellule beta. L’inibizione a breve termine di IGFBP3/TMEM219 in vitro e in vivo e l’ablazione genetica di TMEM219 hanno preservato le cellule beta e hanno impedito/ritardato l’insorgenza del diabete, mentre il blocco a lungo termine di IGFBP3/TMEM219 ha consentito l’espansione delle cellule beta. È interessante notare che in diverse coorti di pazienti il ripristino di livelli appropriati di IGFBP3 è stato associato a una migliore funzione delle cellule beta. Il percorso IGFBP3/TMEM219 ha quindi dimostrato di essere un regolatore fisiologico dell’omeostasi delle cellule beta ed è anche dimostrato che è interrotto nel T1D/T2D. Il target di IGFBP3/TMEM219 può quindi servire come opzione terapeutica nel diabete”.
Fiorina ha fondato una società di biotecnologie in Italia chiamata Enthera nel 2016 che sta sviluppando prodotti biologici basati su queste scoperte. I primi test sull’uomo di un trattamento con anticorpi per bloccare il percorso IGFBP3/TMEM219 potrebbero iniziare già a settembre 2022 in Europa.
Fonte:Nature