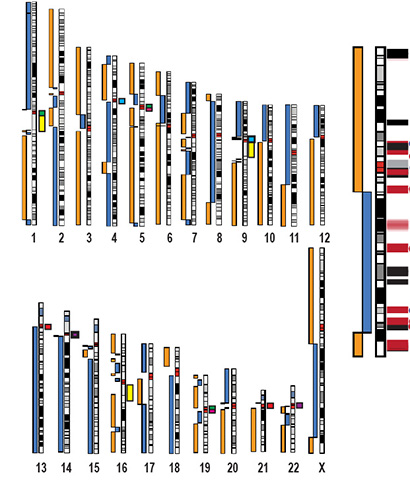
(Cromosoma X-Immagine: deogrammi cromosomici che mostrano l’assemblaggio dell’intero genoma umano CHM13, superando tutti i precedenti assemblaggi del genoma umano in termini di continuità, completezza e accuratezza. Il cromosoma X è ingrandito a destra. I contorni senza pause sono illustrati come barre blu e arancioni accanto agli ideogrammi cromosomici. (Credito: Miga et al., Nature 2020).
Sebbene l’attuale genoma di riferimento umano sia il genoma dei vertebrati più accurato e completo mai prodotto, ci sono ancora lacune nella sequenza del DNA, anche dopo due decenni di miglioramenti. Ora, per la prima volta, gli scienziati hanno determinato la sequenza completa di un cromosoma umano da un’estremità all’altra (“da telomero a telomero”) senza lacune e con un livello di accuratezza senza precedenti.
La pubblicazione dell’assemblaggio telomero-telomero di un cromosoma X umano completo il 14 luglio su Nature è un traguardo fondamentale per i ricercatori di genomica. L’autrice principale dello studio Karen Miga, ricercatrice presso la UC Santa Cruz Genomics Institute, ha affermato che il progetto è stato reso possibile dalle nuove tecnologie di sequenziamento che consentono “letture ultra lunghe”, come la tecnologia di sequenziamento dei nanopori sperimentata presso l’UC Santa Cruz
Le sequenze ripetitive di DNA sono comuni in tutto il genoma e hanno sempre rappresentato una sfida per il sequenziamento perché la maggior parte delle tecnologie produce “letture” relativamente brevi della sequenza, che devono poi essere messe insieme come un puzzle per assemblare il genoma. Le sequenze ripetitive producono molte letture brevi che sembrano quasi identiche, come una grande distesa di cielo blu in un puzzle, senza indizi su come i pezzi si incastrano o quante ripetizioni ci sono.
“Queste sequenze ricche di ripetizioni una volta erano considerate intrattabili, ma ora abbiamo fatto passi da gigante nella tecnologia di sequenziamento”, ha detto Miga. “Con il sequenziamento dei nanopori, otteniamo letture ultra lunghe di centinaia di migliaia di coppie di basi che possono coprire un’intera regione di ripetizione, in modo da aggirare alcune delle sfide”.
Colmare le lacune rimanenti nella sequenza del genoma umano apre nuove regioni del genoma in cui i ricercatori possono cercare associazioni tra variazioni di sequenza e malattia e per altri indizi su importanti questioni sulla biologia umana e l’evoluzione.
“Stiamo iniziando a scoprire che alcune di queste regioni in cui c’erano lacune nella sequenza di riferimento sono in realtà tra le più ricche di variazione nelle popolazioni umane, quindi ci sono mancate molte informazioni che potrebbero essere importanti per la comprensione della biologia umana e malattia “, ha detto Miga.
Strategia di lucidatura
Il passaggio successivo è stata una strategia di lucidatura che utilizza i dati di più tecnologie di sequenziamento per garantire l’accuratezza di ogni base nella sequenza.
“Abbiamo utilizzato un processo iterativo (l’iterazione è l’atto di ripetere un processo con l’obiettivo di avvicinarsi a un risultato desiderato. Ogni ripetizione del processo è essa stessa definita un’iterazione e i risultati di una sono utilizzati come punto di partenza per quella successiva), su tre diverse piattaforme di sequenziamento per perfezionare la sequenza e raggiungere un alto livello di precisione”, ha spiegato Miga. “Gli indicatori unici forniscono un sistema di ancoraggio per le letture ultra lunghe e, una volta ancorate le letture, puoi utilizzare più set di dati per chiamare ciascuna base”.
Il sequenziamento dei nanopori, oltre a fornire letture ultra lunghe, può anche rilevare basi che sono state modificate dalla metilazione, un cambiamento “epigenetico” che non altera la sequenza, ma ha importanti effetti sulla struttura del DNA e sull’espressione genica. Mappando i modelli di metilazione sul cromosoma X, il team è stato in grado di confermare le osservazioni precedenti e rivelare alcune tendenze interessanti nei modelli di metilazione all’interno del centromero.
La nuova sequenza del genoma umano, derivata da una linea cellulare umana chiamata CHM13, colma molte lacune nell’attuale genoma di riferimento, noto come Genome Reference Consortium build 38 (GRCh38).
Il consorzio T2T sta continuando a lavorare per il completamento di tutti i cromosomi CHM13. “È un consorzio aperto, quindi per molti aspetti questo è un progetto guidato dalla comunità, con molte persone che dedicano tempo e risorse ad esso”, ha detto Miga.
Oltre a Miga e Phillippy, gli autori del documento includono il primo autore Sergey Koren presso il National Human Genome Research Institute e scienziati di quasi due dozzine di istituzioni negli Stati Uniti e nel Regno Unito, tra cui l’Università di Washington, la Johns Hopkins University, UC San Diego e il Wellcome Sanger Institute. Questo lavoro è stato sostenuto dal National Institutes of Health degli Stati Uniti.
Da telomero a telomero
Miga e Adam Phillippy del National Human Genome Research Institute (NHGRI), entrambi autori corrispondenti del nuovo articolo, hanno co-fondato il consorzio Telomere-to-Telomere (T2T) per perseguire un assemblaggio completo del genoma dopo aver lavorato insieme su un documento del 2018 che ha dimostrato il potenziale della tecnologia dei nanopori per produrre una sequenza completa del genoma umano. Questo sforzo ha utilizzato il sequencer MinION di Oxford Nanopore Technologies, che sequenzia il DNA rilevando il cambiamento nel flusso di corrente quando singole molecole di DNA passano attraverso un minuscolo foro (un “nanoporo”) in una membrana.
Il nuovo progetto si è basato su questo sforzo, combinando il sequenziamento dei nanopori con altre tecnologie di sequenziamento di PacBio e Illumina e mappe ottiche di BioNano Genomics. Utilizzando queste tecnologie, il team ha prodotto un assemblaggio dell’intero genoma che supera tutti i precedenti assemblaggi del genoma umano in termini di continuità, completezza e accuratezza, superando anche l’attuale genoma di riferimento umano per alcune metriche.
“Tuttavia, c’erano ancora più interruzioni nella sequenza”, ha detto Miga. Per completare il cromosoma X, il team ha dovuto risolvere manualmente diverse lacune nella sequenza. Due duplicazioni segmentali sono state risolte con letture di nanopori ultra lunghe che coprivano completamente le ripetizioni ed erano ancorate in modo univoco su entrambi i lati. La rottura rimanente era al centromero, una regione notoriamente difficile di DNA ripetitivo trovato in ogni cromosoma.
Nel cromosoma X, il centromero comprende una regione di DNA altamente ripetitivo che copre 3,1 milioni di paia di basi (le basi A, C, T e G formano coppie nella doppia elica del DNA e codificano le informazioni genetiche nella loro sequenza). Il team è stato in grado di identificare le varianti all’interno della sequenza di ripetizione che ha utilizzato per allineare le letture lunghe e collegarle insieme per coprire l’intero centromero.
“Per me, l’idea di poter mettere insieme una ripetizione in tandem da 3 megabase è semplicemente strabiliante. Ora possiamo raggiungere queste regioni ripetute che coprono milioni di basi che in precedenza erano ritenute intrattabili “, ha detto Miga.
La prima sequenza di DNA end-to-end di un cromosoma umano è stata ora assemblata. Fino a questo momento, e nonostante decenni di miglioramenti nell’assemblaggio del genoma di riferimento umano, nessun cromosoma umano era mai stato assemblato da inizio alla fine.
In questo consorzio Telomere-to-Telomere, i ricercatori stanno lavorando per assemblare un riferimento più completo dell’intero genoma umano, cromosoma per cromosoma.
Il consorzio prende il nome dai cappucci protettivi che segnano le estremità opposte di ciascun cromosoma. La fonte di finanziamento e centro organizzativo è il National Human Genome Research Institute presso il National Institutes of Health.
I ricercatori hanno iniziato con il cromosoma X, in parte perché un assemblaggio più completo potrebbe scoprire nuove informazioni su molte condizioni mediche legate al sesso.
Le persone di solito hanno 23 cromosomi accoppiati. Generalmente, una femmina biologica ha due cromosomi X, uno per ogni genitore. Un maschio biologico ha ereditato un cromosoma X da sua madre e un cromosoma Y da suo padre.
“Il primo assemblaggio completo da telomero a telomero di un cromosoma umano dimostra che ora potrebbe essere possibile completare l’intero genoma umano utilizzando le tecnologie disponibili”, hanno scritto gli scienziati nel loro articolo. Sebbene si siano concentrati sulla finitura del cromosoma X, hanno anche iniziato a ricostruire altri cromosomi umani.
Mentre alcuni dei restanti cromosomi umani dovrebbero essere relativamente facili da assemblare, i ricercatori hanno notato che molti altri sono difficili. L’area più difficile da esplorare del cromosoma X era il suo centromero. Questa è la regione del connettore altamente condensata per la replicazione cromosomica.
I metodi e i risultati del sequenziamento del cromosoma X sono stati riportati in una versione rapida e precoce di un manoscritto pubblicato il 14 luglio sulla rivista scientifica Nature.
Più di 30 scienziati di diverse istituzioni negli Stati Uniti e all’estero hanno collaborato a questo sforzo. A dirigere la ricerca c’erano l’autore principale e corrispondente Karen H. Miga dell’Università della California di Santa Cruz, l’autore co-responsabile Sergey Koren e l’autore senior Adam Phillippy, entrambi del National Human Genome Research Institute.
Tra gli scienziati che hanno contribuito con la loro esperienza a questo lavoro diversi provenivano dalla University of Washington School of Medicine di Seattle. Tra questi Evan Eichler, Professore di scienze del genoma e ricercatore dell’Howard Hughes Medical Institute, Mitchell R. Vollger, David Porubsky, Glennis A. Logsdon e Amy B. Wilfert, tutti del Dipartimento di Scienze del genoma e Jeanne Fredrickson e Rosa ana Risques, entrambi del Dipartimento di Patologia.
“Questo è un primo passo importante e una realizzazione degli obiettivi a lungo termine del Progetto Genoma Umano”, ha detto Eichler. “L’assemblaggio completo di altri cromosomi e quindi i genomi diploidi completi sono le prossime sfide“.
Il cromosoma X in questo studio proveniva da una mola idatiforme, una crescita eccessiva di tessuto da una placenta. Questo tessuto è speciale perché trasporta solo copie dei 23 cromosomi umani del padre, piuttosto che un set sia della madre che del padre. Questo rende meno complicato analizzare il contenuto dei suoi cromosomi. Il campione di DNA della mola idatiforme è stato originariamente selezionato dal laboratorio Eichler più di 10 anni fa per risolvere complesse regioni genetiche umane che erano rimaste incompiute come parte del Progetto Genoma Umano.
L’ultimo studio ha beneficiato dei progressi nelle letture del DNA ad alta copertura e ultra lunghe. Invece di cercare di capire come mettere insieme molte letture brevi, gli scienziati sono riusciti a trovare pezzi di puzzle più piccoli e più grandi. Il sequenziamento ultra lungo Oxford Nanopore ha reso più veloce, più accurato e meno costoso analizzare il DNA. Questa tecnologia identifica l’ordine delle quattro sostanze chimiche che codificano il DNA attraverso le fluttuazioni di una corrente elettrica mentre passano attraverso un foro molto piccolo. Altre nuove tecnologie hanno aiutato a convalidare i risultati.
Fonte: Comunicato stampa dell’Università della California Santa Cruz –Comunicato stampa del National Human Genome Research Institute