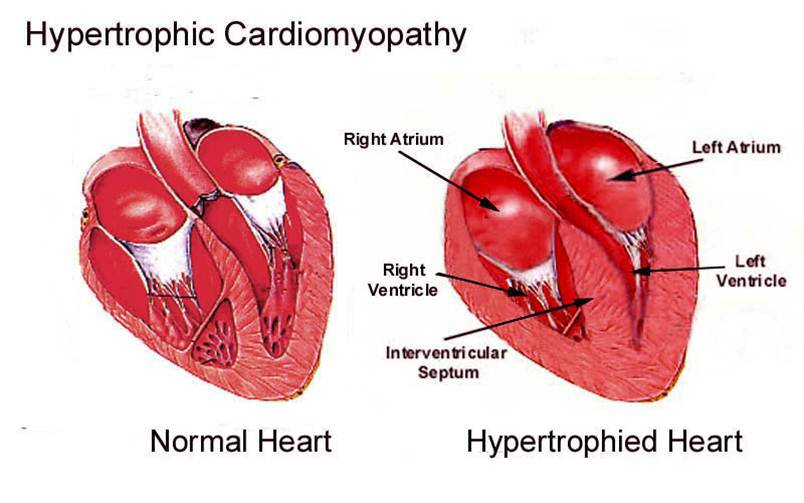
La cardiomiopatia ipertrofica (HCM) è la più comune di tutte le cardiopatie genetiche ed è la principale causa di morte cardiaca improvvisa. È caratterizzata da un ispessimento anomalo del muscolo cardiaco, che nel tempo può portare a disfunzione cardiaca e, infine, insufficienza cardiaca.
In un articolo pubblicato su Proceedings of the National Academy of Sciences ( PNAS ), Beth Pruitt, Prof.ssa di ingegneria meccanica della UC Santa Barbara e Direttrice dell’Institute for BioEngineering del campus, descrive i risultati di una complessa collaborazione a lungo termine che ha coinvolto ricercatori della Stanford University, dell’Università di Washington e dell’Università del Kentucky. Lo studio ha portato a una nuova comprensione di come le mutazioni genetiche si manifestano a livello cellulare per causare la cardiomiopatia ipertrofica e nuove prospettive su come prevenirla.
Nel loro articolo, gli autori spiegano che sono state identificate più di mille mutazioni genetiche che causano l’HCM. La maggior parte di esse si trova nei geni che codificano per le proteine sarcomeriche, i mattoni strutturali del muscolo cardiaco responsabili della generazione e della regolazione della contrazione e del rilassamento. Circa un terzo delle mutazioni si trova nella miosina cardiaca beta, la proteina primaria che guida la contrazione delle cellule cardiache. La contrazione del muscolo cardiaco e di ogni altro muscolo del nostro corpo, deriva da un processo in cui la proteina miosina “cammina” lungo una catena di molecole di actina, un processo noto come ciclo del ponte incrociato. Durante questo processo, l’energia chimica sotto forma di ATP viene convertita in energia meccanica, portando infine alla contrazione cardiaca.
Prima di una contrazione, la testa di un filamento di una molecola di miosina a due filamenti intrecciati è rimboccata contro una molecola di actina. La contrazione muscolare inizia quando una molecola di ATP, nota come “moneta energetica” dei sistemi biologici, si lega alla testa della miosina. La testa della miosina e l’ATP attaccato si staccano quindi dall’actina, avviando l’idrolisi dell’ATP, che viene trasformato in ADP più un gruppo fosfato. Questo processo rilascia energia che “arma” la proteina miosina in uno stato ad alta energia e cambia la forma della miosina in modo che sia pronta a strisciare lungo l’actina. A quel punto, il fosfato viene rilasciato dalla miosina, facendo sì che la miosina spinga sull’actina e rilasci il fosfato, che porta la miosina a camminare verso la catena successiva di actina e a contrarre il muscolo.
Poiché la cardiomiopatia ipertrofica è spesso osservata in pazienti con mutazioni nella proteina beta miosina cardiaca, è stato ipotizzato che le mutazioni dell’HCM causino una cascata di eventi che si manifestano, in ultima analisi, in un danno al cuore stesso. Questo studio ha messo alla prova questa idea, concentrandosi su una singola mutazione, P710R, che ha drasticamente ridotto la velocità di motilità in vitro, la velocità con cui il motore della miosina cammina sull’actina, in contrasto con altre mutazioni MYH7, che hanno portato ad un aumento della velocità di motilità.
Il principale obiettivo di questo progetto di ricerca era capire come una mutazione legata alla malattia cardiaca nei pazienti modifica la funzione cardiaca a livello cellulare.
Il team ha utilizzato la tecnologia CRISPR per modificare i cardiomiociti di cellule staminali pluripotenti indotte umane (cellule responsabili della contrazione cardiaca) inserendo in essi la mutazione P710R. Pruitt guida la banca di cellule staminali dell’UCSB, dove vengono mantenute e riprodotte linee cellulari “pulite”, prive di anomalie genetiche, per i ricercatori universitari. Tali linee pulite e prive di mutazioni forniscono un punto di riferimento perfetto per il confronto con le cellule per vedere in modo molto preciso gli effetti della mutazione P710R. Ad esempio, il team di ricerca sta ora testando gli effetti di diverse mutazioni legate a malattie cardiache nello stesso background genetico.
Vedi anche:Cardiomiopatia ipertrofica, gli scienziati catturano l’interruttore molecolare heartbeat in azione
“Puoi avere dieci persone con la stessa mutazione genetica in questa proteina e possono avere vari gradi di significato clinico, perché il resto del loro genoma è diverso; questo è ciò che ci rende individui”, ha detto Pruitt. “Queste linee ci consentono di esaminare quale sia il risultato della mutazione genetica. Confrontando l’effetto di diverse mutazioni, possiamo iniziare a distinguere come questi cambiamenti portano alla cardiomiopatia ipertrofica. Ci consente di osservare da vicino come e perché le cellule si adattano alla mutazione in quel modo e ottenere dati e metterli in relazione con lo spessore della parete del cuore e tutte le altre cose che accadono a valle”.
Questa ricerca è iniziata quasi 15 anni fa, mentre Pruitt era ancora alla Stanford e ha portato a questo documento collaborativo. Ora la tecnologia CRISPR consente ai ricercatori di progettare cellule che esprimono mutazioni specifiche collegate a malattie cardiache e quindi valutare i cambiamenti molecolari e funzionali per determinare l’impatto cellulare delle singole mutazioni che sono state identificate nei pazienti con HCM. Questi studi forniranno una comprensione meccanicistica di come le singole mutazioni a livello molecolare si traducano in cardiomiopatia ipertrofica nei pazienti.
In questo progetto, una volta introdotta la mutazione, le cellule sono state saggiate in collaborazione tra il Pruitt lab (UCSB) e il Bernstein lab (Stanford University), utilizzando la microscopia a forza di trazione, un saggio che consente l’osservazione simultanea di una cellula battente e della forza che genera. Il laboratorio Spudich (Stanford), ha condotto studi separati sulla stessa proteina mutata a livello molecolare utilizzando una trappola ottica, in cui viene applicata una leggera pressione per controllare con precisione la posizione e la forza di un “manubrio” di actina tenuto tra le perline mentre le teste di miosina camminano lungo l’actina, per misurare il ciclo di alimentazione della miosina. Il test ha rivelato che la mutazione P710R ha ridotto la dimensione del passo del motore della miosina (cioè la lunghezza di ogni passo) e la velocità con cui la miosina si stacca dall’actina.
In una collaborazione con il ricercatore dell’Università del Kentucky Kenneth Campbell, queste osservazioni sono state poi confrontate con un modello computazionale di come i motori della miosina interagiscono nella cellula per generare forza. I risultati hanno confermato un ruolo chiave per la regolazione di quello che viene chiamato “stato super-rilassato” della miosina. Come ha spiegato Pruitt, le teste di miosina trascorrono molto tempo in uno stato super-rilassato, riferendosi a quando non è legata all’actina. Qualsiasi mutazione o farmaco che cambia per quanto tempo o quanto fortemente i motori della miosina sono legati all’actina, cambierà la forza cellulare e modificherà gli eventi di segnalazione a valle che guidano il rimodellamento e la crescita o l’ipertrofia.
La mutazione P710R in questo studio è stata trovata per destabilizzare lo stato super-rilassato. Di conseguenza, più teste di miosina sono legate all’actina nelle cellule che ospitano la mutazione, il che spiega l’aumento di forza osservato in quelle cellule.
Ha affermato Pruitt: “E’ davvero fenomenale testare direttamente come una particolare mutazione introduce cambiamenti che portano alla HCM. Possiamo capire cosa succede a livello cellulare. Quindi possiamo iniziare a sviluppare modelli e identificare terapie farmacologiche di prossima generazione. Invece di identificare solo i sintomi, possiamo guardare i meccanismi che sono alla base delle disfunzioni e poi affrontare quelle a livello cellulare prima che si trasformino in una malattia”.
Fonte: PNAS