(Alzheimer-Immagine Credit Public Domain).
Il morbo di Alzheimer (AD) è una malattia neurodegenerativa che colpisce milioni di persone in tutto il mondo, per la quale sono attualmente disponibili solo trattamenti sintomatici. I meccanismi esatti alla base dell’AD rimangono poco chiari nonostante i tentativi di comprenderne la fisiopatologia.
La teoria più importante postula che, nell’AD, le proteine tau e Aβ influenzino negativamente le cellule neuronali compromettendo l’approvvigionamento energetico e la risposta antiossidante, causando disfunzione mitocondriale e sinaptica. L’attività neuronale è altamente dipendente dall’energia e i neuroni sono particolarmente sensibili all’interruzione della funzione mitocondriale. Inoltre, i mitocondri producono energia cellulare (adenosina trifosfato; ATP) e sono anche coinvolti in molti processi importanti per la vita e la morte della cellula, compreso il controllo dei livelli del secondo messaggero, come gli ioni calcio (Ca 2+ ) e specie reattive dell’ossigeno (ROS).
È importante sottolineare che la disfunzione mitocondriale contribuisce a ridurre la produzione di ATP, Ca 2+disomeostasi e generazione di ROS. Alterazioni nella dinamica mitocondriale e nella mitofagia si verificano nella fase iniziale dell’AD, ma i meccanismi sottostanti sono poco conosciuti.
Pertanto, gli studi che chiariscono i meccanismi delle anomalie mitocondriali nell’AD faciliteranno una maggiore comprensione della patogenesi di questa malattia neurodegenerativa e potenzialmente contribuiranno al progresso di strategie terapeutiche per proteggere l’attività sinaptica e la successiva funzione cognitiva.
“Esaminiamo gli studi che suggeriscono un ruolo della disfunzione mitocondriale e la conseguente produzione di ROS nella patologia di AD e forniscono un contesto per spiegare gli approcci terapeutici attuali e futuri. Suggeriamo che il miglioramento della funzione mitocondriale dovrebbe essere considerato un importante intervento terapeutico contro l’AD”, spiega Afzal Misrani, ricercatore post-dottorato presso l’Università di Guangzhou Canton, Cina che ha collaborato con Sidra Tabassum e Li Yang, ricercatori post-dottorato presso la stessa Università.
Disfunzione mitocondriale nella malattia di Alzheimer
I mitocondri svolgono un ruolo fondamentale nella bioenergetica e nelle funzioni respiratorie, che sono essenziali per i numerosi processi biochimici alla base della vitalità cellulare. La morfologia mitocondriale cambia rapidamente in risposta a insulti esterni e cambiamenti nello stato metabolico tramite processi di fissione e fusione (la cosiddetta dinamica mitocondriale) che mantengono la qualità mitocondriale e l’omeostasi. I mitocondri danneggiati vengono rimossi mediante un processo noto come mitofagia, che comporta la loro degradazione da parte di una specifica via autofagosomica.
Negli ultimi anni sono stati compiuti notevoli sforzi per studiare l’impatto sulla patogenesi della malattia di Alzheimer (AD) di varie forme di disfunzione mitocondriale, come la produzione eccessiva di specie reattive dell’ossigeno (ROS), Ca 2+ mitocondrialedysomeostasi, perdita di ATP e difetti nella dinamica e nel trasporto mitocondriale e mitofagia.
Ricerche recenti suggeriscono che il ripristino della funzione mitocondriale mediante l’esercizio fisico, una dieta antiossidante o approcci terapeutici possono ritardare l’insorgenza e rallentare la progressione dell’AD.
“In questa recensione, ci concentriamo sui recenti progressi che evidenziano il ruolo cruciale delle alterazioni nella funzione mitocondriale e dello stress ossidativo nella patogenesi dell’AD, sottolineando un quadro di approcci terapeutici esistenti e potenziali”, spiegano gli autori.
Fig.1
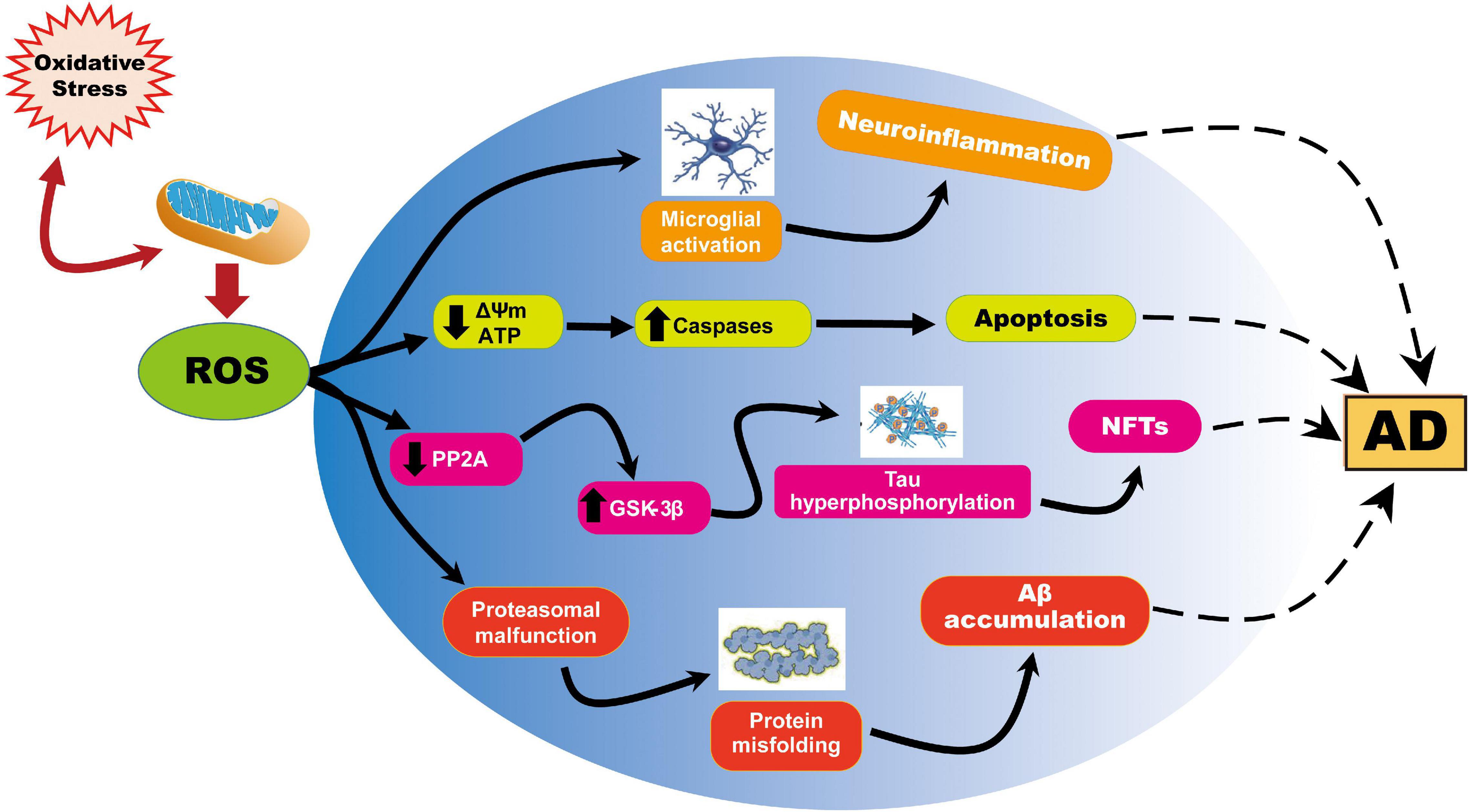
Immagine: rappresentazione delle anomalie mitocondriali indotte da ROS nell’AD. La produzione di ROS o il sistema antiossidante compromesso provoca l’equilibrio redox cellulare in uno squilibrio ossidativo e causa la sovrapproduzione di ROS. I ROS generati durante la respirazione cellulare hanno effetti dannosi sui mitocondri e sulla funzione neuronale. L’aumento di ROS provoca la riduzione della generazione mitocondriale di ΔΨm e di ATP influenzando negativamente le riserve di energia mitocondriale, il disturbo del metabolismo energetico e la dinamica e la mitofagia compromesse. ROS provoca inoltre un aumento dell’attività della caspasi e avvia l’apoptosi. D’altra parte, la sovrapproduzione di ROS provoca l’inibizione della fosfatasi 2A (PP2A), che attiva anche la glicogeno sintasi chinasi (GSK) 3β causando iperfosforilazione della tau e accumulo di grovigli neurofibrillari.
Fig.2
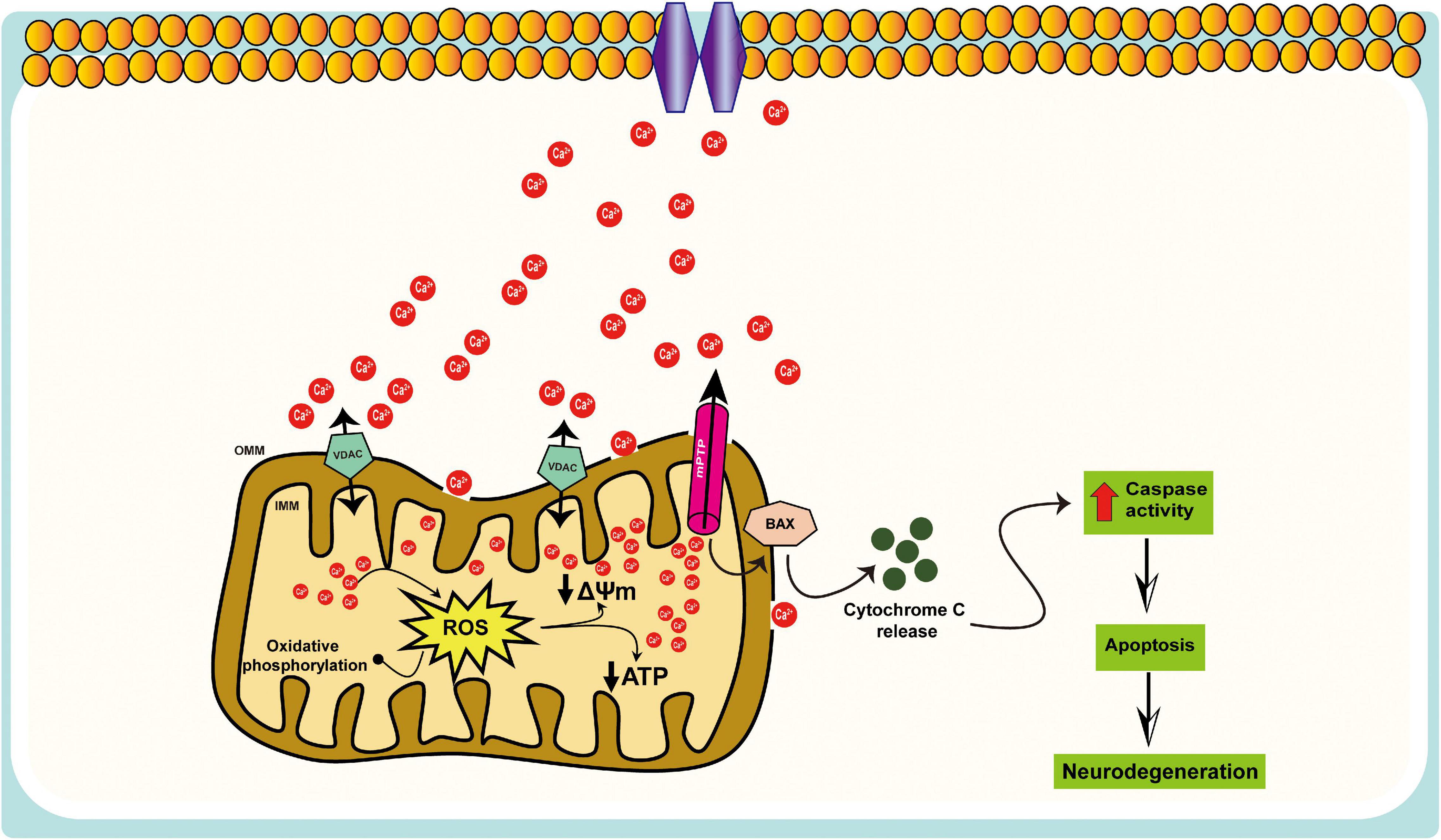
Immagine: rappresentazione schematica della disregolazione mitocondriale del Ca 2+ nell’AD. I mitocondri partecipano alla segnalazione intracellulare di Ca 2+ come modulatori, tamponi e sensori; un eccesso di Ca 2+ assorbito dai mitocondri può portare alla morte cellulare, cioè un sovraccarico di Ca 2+ mitocondriale, provoca un aumento della produzione di ROS, inibizione della sintesi di ATP, apertura dei pori di transizione della permeabilità mitocondriale (mPTP), rilascio del citocromo c, attivazione delle caspasi e l’apoptosi.
Vedi anche: Alzheimer: bassi livelli di beta amiloide la causa?
L’analisi di cui sopra chiarisce che sono necessarie strategie in grado di prendere di mira la funzione mitocondriale per rallentare la progressione dell’AD. L’obiettivo dello sforzo di ricerca dovrebbe essere quello di sviluppare un intervento terapeutico che possa prendere di mira i ROS e l’eccessiva frammentazione mitocondriale, riducendo così al minimo la disfunzione mitocondriale e il conseguente danno sinaptico durante la progressione dell’AD (riassunto nella Tabella 1).
Fonte: Frontiers in neuroscience