
Neurodegenerazione-Immagine: in primo piano, le fibrille TDP-43 comprendenti proteine a lunghezza intera vengono digerite dall’enzima Proteinasi K (da sinistra a destra), assottigliando la fibrilla esponendo il nucleo amiloide.
Gli scienziati dell’EPFL hanno riprodotto le caratteristiche chiave degli aggregati proteici patologici trovati nel cervello di pazienti con malattia di Lou Gehrig e altre malattie neurologiche, fornendo informazioni sul meccanismo sottostante e offrendo strade promettenti per nuove terapie.
I risultati sono stati pubblicati su Nature Neuroscience.
Diverse malattie neurodegenerative, come l’Alzheimer, il Parkinson e la malattia di Lou Gehrig, nota anche come sclerosi laterale amiotrofica (SLA), sono causate da proteine che si smarriscono e iniziano ad aggregarsi in fibrille che si accumulano in specifiche regioni del cervello. Ora, gli scienziati dell’EPFL hanno scoperto un nuovo meccanismo che spiega come gli aggregati diventano patologici e si diffondono in diverse regioni del cervello. Uno dei principali sospettati è la proteina altamente instabile chiamata TDP43. Gli scienziati hanno scoperto che gli aggregati TDP43 che si formano nel cervello non sono implicitamente patogeni fino a quando non vengono elaborati per rivelare il loro nucleo “appiccicoso”.
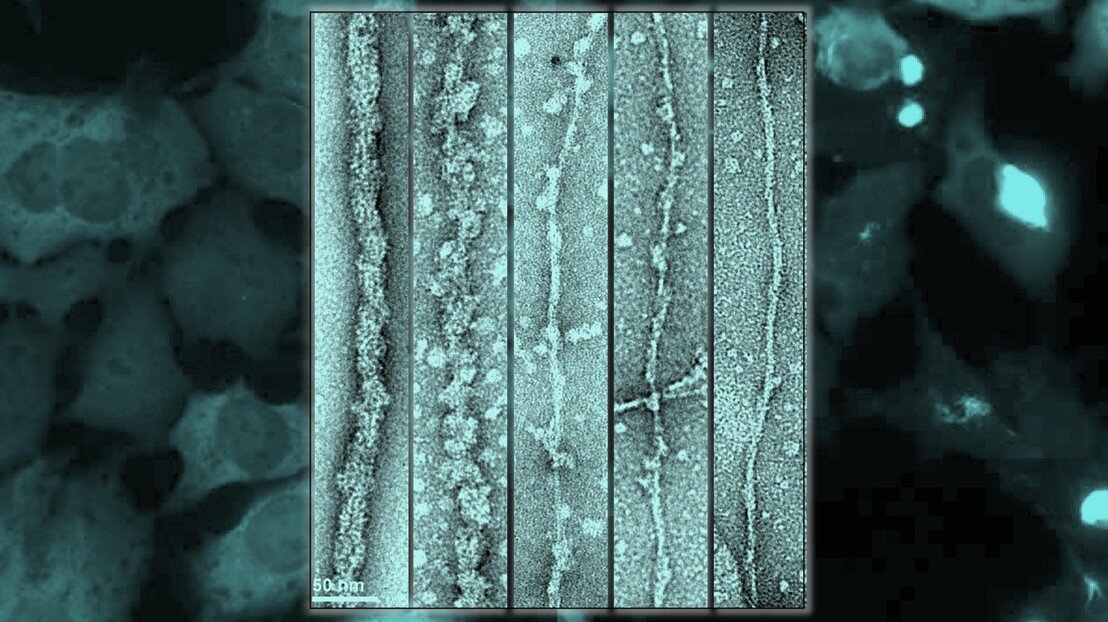
Immagine:come l’esposizione del nucleo della fibrilla TDP-43 guida la patologia la nella cellula. Credito: EPFL / Galina Limorenko.
L’aggregazione della proteina TDP43 è un segno distintivo della SLA e di altre malattie neurodegenerative. Una volta formati, gli aggregati di TDP43 possono diffondersi in diverse regioni del cervello dove corrompono il TDP-43 normale e funzionale. Ma cosa fa scattare l’aggregazione TDP-43 in primo luogo? Quali sono i meccanismi responsabili dello scatenamento dei suoi effetti patogeni? Questa lacuna di conoscenza ostacola lo sviluppo di farmaci efficaci per bloccare l’aggregazione del TDP-43 o neutralizzarne le proprietà tossiche.
Scatenare gli effetti patogeni di TDP43 mediante scissione
In questo ultimo studio dell’EPFL condotto in collaborazione con gli scienziati dell’Università della Pennsylvania, il Dr. Senthil Kumar e il Prof. Hilal Lashuel hanno scoperto un nuovo meccanismo responsabile degli effetti patogeni dell’aggregato TDP43, preparato nella provetta o isolato dai cervelli dei pazienti post-mortem. Le superfici di questi aggregati TDP43 devono prima essere scisse dagli enzimi per rivelare superfici appiccicose nascoste che attraggono le normali proteine TDP-43 e inducono la formazione di più aggregati.
“La scoperta è stata facilitata dalla nostra capacità di sviluppare un nuovo metodo per la produzione di fibrille, in laboratorio, che condividono caratteristiche morfologiche e strutturali con quelle riscontrate nel cervello di pazienti affetti da SLA”, afferma il Dott. Senthil T. Kumar, primo autore dell’articolo.
, assottigliando la fibrilla esponendo il nucleo amiloide. Sullo sfondo, il modello cellulare mostra la semina da parte delle fibrille solo quando il nucleo è esposto: sul lato destro, le macchie luminose, mentre quando il nucleo della fibrilla è mascherato, a sinistra, non c'è semina degli aggregati nelle cellule , quindi possiamo vedere solo la colorazione diffusa. Ciò dimostra che il nucleo amiloide deve essere smascherato per rendere le fibrille tossiche e capaci di seminare. Credito: EPFL / Galina Limorenko")
Usando la microscopia crioelettronica, dove i campioni vengono congelati criogenicamente prima di essere osservati attraverso un microscopio elettronico, i ricercatori hanno dimostrato che i filamenti TDP-43 sono sepolti all’interno di un filamento più grande e sono inaccessibili, cioè non ancora patologici, perché sono coperti parti della proteina. Finché questi filamenti sono sepolti, esistono in modalità invisibile e non sono accessibili ad altre molecole o proteine. In altre parole, il TDP43 diventa patologico quando il suo rivestimento esterno viene tagliato per rivelare i suoi filamenti interni “appiccicosi”, ma rimane in modalità invisibile quando il suo rivestimento esterno è intatto.
“I nostri risultati suggeriscono che l’inibizione degli enzimi responsabili della scissione del filamento TDP-43 rappresenta una valida strategia terapeutica per rallentare la formazione di aggregati TDP-43 e prevenirne la diffusione nel cervello, rallentando così la progressione della malattia. Come passo successivo, abbiamo in programma di identificare questi enzimi e determinare se l’inibizione della loro attività potrebbe prevenire l’aggregazione e la neurodegenerazione di TDP-43 in modelli cellulari e animali di SLA“, afferma Hilal Lashuel, Professore dell’EPFL che gestisce il laboratorio che ha condotto lo studio.
I nuovi risultati hanno implicazioni anche per lo sviluppo di nuovi strumenti e metodi per la diagnosi precoce della SLA e di altre malattie neurodegenerative. Lo strato globulare protettivo potrebbe spiegare perché le fibrille TDP-43 sono così difficili da rilevare. I metodi e i coloranti standard comunemente usati per rilevare e monitorare la formazione di fibrille da parte di altre proteine sospette nel cervello spesso non sono riusciti a rilevare le fibrille TDP-43. “Spiega anche perché è stato molto difficile sviluppare agenti di imaging utilizzando fibrille TDP-43 intatte. Tali agenti di imaging sono assolutamente necessari per consentire la diagnosi precoce, monitorare la progressione della malattia e valutare l’efficacia di nuove terapie”, afferma il Dott. Kumar.
Vedi anche:Octopamina: il segnale SOS del cervello nella neurodegenerazione
Importanza di studiare la proteina a lunghezza intera
TDP-43 è una proteina altamente instabile e si aggrega rapidamente in diverse strutture, rendendo così difficile generare aggregati di TDP-43 simili a patologie in modo riproducibile. Ciò ha costretto molti scienziati a lavorare con piccoli frammenti della proteina, in particolare frammenti della regione responsabile della sua aggregazione. “Quando abbiamo determinato la struttura del frammento proteico che forma il nucleo delle fibrille TDP-43 preparate in laboratorio, abbiamo ottenuto una struttura diversa da quella delle fibrille TDP-43 isolate dal cervello di un paziente, anche se la sequenza aminoacidica di questi frammenti è praticamente identica“, afferma il Dott. Kumar.
“I nostri risultati dimostrano che le sequenze proteiche che fiancheggiano questa regione soggetta ad aggregazione svolgono un ruolo importante nel dettare la struttura finale e che la riproduzione delle proprietà degli aggregati TDP-43 nel cervello richiede di lavorare con la proteina a lunghezza intera”, afferma Hilal Lashuel. “Questo è essenziale per garantire che i farmaci, gli anticorpi e gli agenti di imaging che sviluppiamo in laboratorio abbiano maggiori possibilità di coinvolgere gli aggregati TDP-43 rilevanti per la malattia nel cervello dei pazienti”.
I ricercatori hanno dimostrato di poter produrre fibrille TDP-43 con la stessa sequenza centrale di quella delle fibrille dal cervello dei pazienti. “Ma dobbiamo ancora determinare se il nucleo della fibrilla non mascherato ha la stessa struttura“, chiarisce Hilal Lashuel.
“Se lo mostriamo, allora avremo l’unico sistema che consente di produrre la patologia effettiva nella provetta. Ciò avrà enormi implicazioni per comprendere come le mutazioni legate alla malattia e le modificazioni proteiche influenzino l’aggregazione del TDP-43 e faciliterebbero la sviluppo di nuovi farmaci che bloccano l’aggregazione del TDP-43, ne neutralizzano la patogenicità o si legano agli aggregati del TDP-43 e ne facilitano il rilevamento nel cervello”.
Fonte:Nature