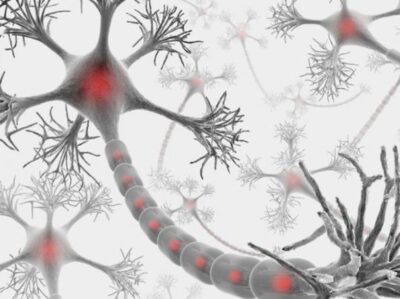
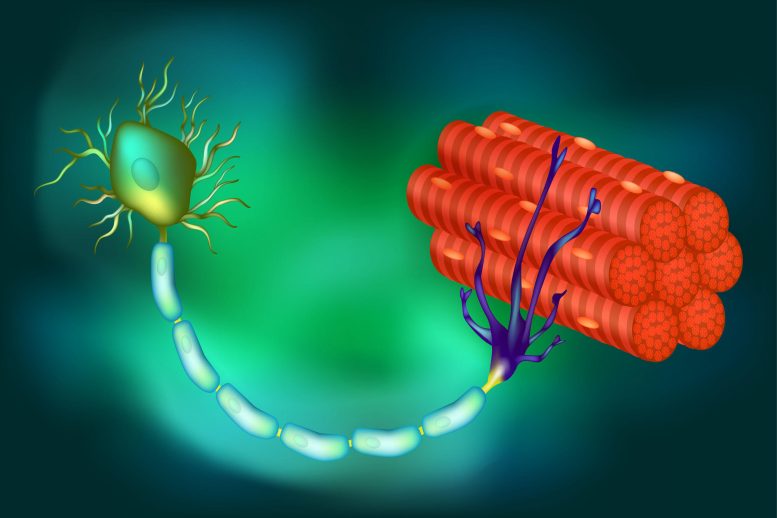
Un’illustrazione delle connessioni tra le fibre muscolari e un motoneurone, l’ultimo dei quali si basa su proteine cruciali per creare e inviare segnali alle fibre per contrarsi, con conseguente movimento. In alcune malattie, come la SLA, queste proteine vengono perse, i neuroni muoiono e ne risulta la paralisi. Credito: Centro di movimento somatico
Nella ricerca che ha coinvolto sia i topi che gli esseri umani, è stato scoperto che un farmaco geneticamente modificato reintegra i livelli di una proteina critica, preservando così la funzione dei motoneuroni. Questa svolta, particolarmente rilevante per la sclerosi laterale amiotrofica in cui questa funzione è tipicamente compromessa, potrebbe potenzialmente aprire la strada a studi clinici.
In quasi tutti i casi di sclerosi laterale amiotrofica (SLA) e in almeno la metà di tutti i casi di malattia di Alzheimer (AD) e demenza frontotemporale, una proteina nota come TDP-43 è fuori luogo rispetto alla sua posizione abituale nel nucleo della cellula. Questo spostamento comporta la perdita di stathmin-2, una proteina vitale per la rigenerazione dei neuroni e la conservazione dei loro legami con le fibre muscolari, entrambi essenziali per la contrazione e il movimento muscolare.
In uno studio pubblicato sulla rivista Science, un gruppo di ricercatori, guidati da Don Cleveland, Ph.D., Distinguished Professor of Medicine, Neurosciences and Cellular and Molecular Medicine presso la University of California San Diego School of Medicine, rivela che la perdita di stathmin-2 può essere invertita utilizzando farmaci a base di DNA appositamente progettati che ripristinano la normale elaborazione dell’RNA che codifica per le proteine.

Cleveland è ampiamente accreditato per aver sviluppato il concetto di farmaci DNA di design, che agiscono per attivare o disattivare i geni associati a molte malattie degenerative dell’invecchiamento del sistema nervoso umano, tra cui SLA, AD, malattia di Huntington e cancro.
Diversi farmaci a DNA di marca sono attualmente in sperimentazione clinica per molteplici malattie. Uno di questi farmaci è stato approvato per il trattamento di una malattia neurodegenerativa infantile chiamata atrofia muscolare spinale.
Il nuovo studio si basa sulla ricerca in corso di Cleveland e altri per quanto riguarda il ruolo e la perdita di TDP-43, una proteina associata a SLA, AD e altri disturbi neurodegenerativi. Nella SLA, la perdita di TDP-43 ha un impatto sui motoneuroni che innervano e innescano la contrazione dei muscoli scheletrici, provocandone la degenerazione, che alla fine porta alla paralisi.
“In quasi tutti i casi di SLA, c’è aggregazione di TDP-43, una proteina che funziona nella maturazione degli intermedi dell’RNA che codificano molte proteine. La ridotta attività del TDP-43 causa un errato assemblaggio della stathmin-2 che codifica l’RNA, una proteina necessaria per il mantenimento della connessione dei motoneuroni ai muscoli”, ha affermato Cleveland. “Senza stathmin-2, i motoneuroni si disconnettono dal muscolo, guidando la paralisi che è caratteristica della SLA. Quello che ora abbiamo scoperto è che possiamo imitare la funzione del TDP-43 con un farmaco di design DNA, ripristinando così il corretto livello di stathmin-2 RNA e proteine nel sistema nervoso dei mammiferi”.
Vedi anche:SLA: individuato marcatore neurochimico collegato alla perdita della funzione motoria
In particolare, i ricercatori hanno modificato i geni nei topi per contenere sequenze del gene umano STMN2 e poi hanno iniettato oligonucleotidi antisenso – piccoli frammenti di DNA o RNA che possono legarsi a specifiche molecole di RNA, bloccando la loro capacità di produrre una proteina o cambiando il modo in cui vengono assemblati i loro RNA finali, nel liquido spinale cerebrale. Le iniezioni hanno corretto l’elaborazione errata del pre-mRNA di STMN2 e ripristinato l’espressione della proteina stathmin-2 completamente indipendente dalla funzione TDP-43.
“I nostri risultati gettano le basi per una sperimentazione clinica per ritardare la paralisi nella SLA mantenendo i livelli di proteina stathmin-2 nei pazienti che utilizzano il nostro farmaco di design DNA”, ha affermato Cleveland.
Fonte:Science