Sindrome di Usher di tipo 1F-Immagine Credit Wikipedia-
La sindrome di Usher di tipo 1F è una malattia genetica rara, ma grave che causa sordità, mancanza di equilibrio e cecità progressiva.
Ora, un team guidato da ricercatori della Harvard Medical School, del Massachusetts Eye and Ear e della Ohio State University ha compiuto un importante primo passo verso lo sviluppo di una terapia genica per la malattia.
La ricerca, condotta sui topi modello della Sindrome di Usher di tipo 1F, è descritta il 26 aprile su Nature Communications .
Gli scienziati hanno progettato un “mini-gene”, una versione abbreviata di un gene, per sostituire il gene mutato in Usher 1F. La mutazione rende le cellule ciliate all’interno dell’orecchio interno incapaci di produrre una proteina chiave coinvolta nella trasmissione del suono. Nei topi, il mini-gene ha aumentato la produzione della proteina mancante, consentendo alle cellule ciliate di percepire il suono e ripristinare l’udito.
Poiché la perdita della vista nella Sindrome di Usher di tipo 1F coinvolge una forma leggermente diversa della stessa proteina, i ricercatori affermano che lo stesso approccio può essere utile per prevenire la cecità.
“I pazienti con Usher 1F nascono con una perdita dell’udito profonda e una perdita progressiva della vista, e finora siamo stati in grado di offrire pochissime soluzioni a queste famiglie“, ha affermato il co-autore senior Artur Indzhykulian, assistente Professore di otorinolaringoiatria della HMS e chirurgia a Mass Eye and Ear.
I ricercatori hanno in programma di continuare a testare il mini-gene in altri modelli animali e, infine, sperano di testarlo sugli esseri umani.
“È completamente devastante nascere sordi e poi perdere la vista, quindi speriamo che questo mini-gene possa alla fine essere trasformato in un trattamento per questa malattia”, ha detto il co-autore senior David Corey, Professore Bertarelli di Scienze mediche traslazionali a l’Istituto Blavatnik dell’HMS.
Applicare le competenze a un nuovo problema
I bambini con la sindrome di Usher nascono in genere completamente sordi o con udito gravemente compromesso, mancanza di equilibrio e perdita della vista nel tempo a causa del deterioramento della retina. La cecità si verifica comunemente dall’età adulta.
Questi problemi sorgono a causa di una mutazione che interferisce con la produzione di una proteina chiamata protocaderina-15, che ha forme leggermente diverse nell’orecchio e nell’occhio ed è necessaria per il corretto funzionamento delle cellule del sistema uditivo e visivo.
I ricercatori del laboratorio Corey sono da tempo interessati al ruolo della protocaderina-15 nell’orecchio interno. Nello specifico, hanno voluto sapere in che modo la proteina aiuta i recettori sensoriali chiamati cellule ciliate nell’orecchio a convertire le vibrazioni dall’ambiente in segnali elettrici, che il cervello interpreta come suono.
Il team di Corey aveva precedentemente scoperto come la protocaderina-15 collabora con un’altra proteina, la caderina 23, nelle cellule ciliate per creare filamenti che aprono fisicamente i canali ionici mentre i fasci vibrano, consentendo alla corrente elettrica di entrare nelle cellule. In assenza di questa proteina, la corrente elettrica non può entrare nelle cellule ciliate, la conversione da vibrazione a elettricità non avviene e il cervello non è in grado di rilevare i suoni.
Attraverso questo lavoro, Corey si interessò alla progettazione di una terapia genica per Usher 1F. La terapia introdurrebbe il DNA che codifica per la protocaderina-15 in una cellula, consentendo alla cellula di iniziare a produrre la proteina.
Tuttavia, la protocaderina-15 è così grande che il suo DNA è troppo grande per la tipica capsula virale utilizzata per trasportare materiale genetico in una cellula. Così i ricercatori hanno deciso di esplorare un’altra opzione: accorciare il DNA per creare un mini-gene che ancora codifica per proteine funzionali, ma è abbastanza piccolo da entrare nella capsula virale.
Un gene diventa un mini-gene
Il primo passo ha comportato la mappatura scrupolosa di tutti i 25.000 atomi nella struttura esterna della protocaderina-15 dell’orecchio interno, un processo eseguito dal co-autore senior Marcos Sotomayor, ex ricercatore presso HMS e ora Professore associato di chimica e biochimica presso l’Ohio State.
Usando una combinazione di cristallografia a raggi X e microscopia crioelettronica, Sotomayor ha scoperto che la proteina è composta da atomi disposti in quelli che sembrano 11 anelli di una catena.
Sotomayor ha realizzato otto diverse versioni di protocaderina-15, ciascuna con diversi collegamenti mancanti per rendere la proteina più piccola. I ricercatori hanno quindi decodificato le strutture proteiche troncate in progetti di DNA che hanno potuto testare come mini-geni.
“La conoscenza che abbiamo acquisito studiando la struttura della protocaderina-15 in dettagli ci ha permesso di progettare più rapidamente versioni più brevi della proteina per la terapia genica“, ha spiegato Sotomayor.
Indzhykulian ha testato gli otto mini-geni sulle cellule dell’orecchio interno in una piastra di laboratorio. Ha confermato che le versioni troncate della protocaderina-15 prodotte dal DNA del mini-gene si legano alla caderina 23, la sua partner proteica nelle cellule ciliate.
Da lì, i ricercatori hanno selezionato i tre mini-geni che erano abbastanza piccoli da entrare nella capsula virale.
L’autrice principale dello studio, Maryna Ivanchenko, istruttrice di neurobiologia presso HMS, ha ampiamente testato i tre mini-geni nelle orecchie dei topi che sono stati geneticamente modificati per interrompere la produzione di protocaderina-15. Alla fine, solo un mini-gene ha funzionato.
Il gene ha spinto con successo le cellule ciliate a creare una versione mini della protocaderina-15, che si legava alla caderina-23 e formava i filamenti necessari per aprire i canali ionici. Le cellule ciliate hanno convertito con successo le vibrazioni in segnali elettrici.
I test uditivi sui topi che hanno ricevuto il mini-gene hanno mostrato che i loro cervelli potevano ricevere il segnale sonoro proveniente dalle loro orecchie: gli animali precedentemente sordi potevano sentire.
“Siamo rimasti tutti piacevolmente sorpresi”, ha detto Corey. “Pensavamo che ci sarebbero voluti anni per ottimizzare e provare le cose e modificare la struttura proteica, ma questa versione ha praticamente funzionato”.
“I risultati sono stati entusiasmanti per noi”, ha aggiunto Ivanchenko. “L’aspetto più eccitante delle nostre scoperte è stato che i topi che erano stati completamente sordi ora potevano sentire quasi come i topi normali“.
Dall’orecchio all’occhio
Mentre il mini-gene ha trattato con successo la sordità nel modello murino di Usher 1F, i ricercatori sono ancora più interessati al suo potenziale per il trattamento della cecità associata alla sindrome.
“Purtroppo i bambini con Usher 1F nascono profondamente sordi e potrebbero non avere cellule ciliate nell’orecchio interno ed è improbabile che il mini-gene possa migliorare il loro udito”, hanno affermato gli autori. “Inoltre, molti di questi bambini possono ricevere impianti cocleari che consentono loro di sentire“.
“La cecità è una storia diversa“, hanno osservato i ricercatori, “perché i bambini con Usher 1F nascono con una vista normale. Se il mini-gene potesse produrre la forma di protocaderina-15 mancante nella retina, potrebbe arrestare la perdita della vista”.
Vedi anche:La scoperta dei geni essenziali per la vita aiuta la ricerca sulle malattie rare
Perché iniziare testando il mini-gene nell’orecchio interno del topo se il trattamento della perdita della vista è l’obiettivo principale?
“Principalmente per ragioni logistiche”, hanno detto i ricercatori. “La mancanza di protocaderina-15 causa solo una lieve perdita della vista nei topi e progredisce lentamente. Ciò significa che ci vorrebbero anni per testare i mini- geni nei modelli murini e sarebbe difficile dire quanto bene funzionino. Al contrario, i topi sono nati profondamente sordi, quindi i ricercatori hanno ottenuto risultati chiari entro un paio di settimane“.
“L‘intero progetto è stato progettato per studiare l’orecchio con l’idea che qualcosa che funziona nell’orecchio possa essere successivamente applicato all’occhio”, ha detto Corey. “Mentre il miglior sistema di test è l’orecchio interno del topo, l’obiettivo immediato è un trattamento per la cecità“.
Il laboratorio Corey sta ora testando il mini-gene negli occhi dei pesci zebra, un modello migliore perché questi pesci subiscono una perdita della vista più grave e rapida rispetto ai topi quando la protocaderina-15 non viene prodotta nella retina.
Astratto grafico:
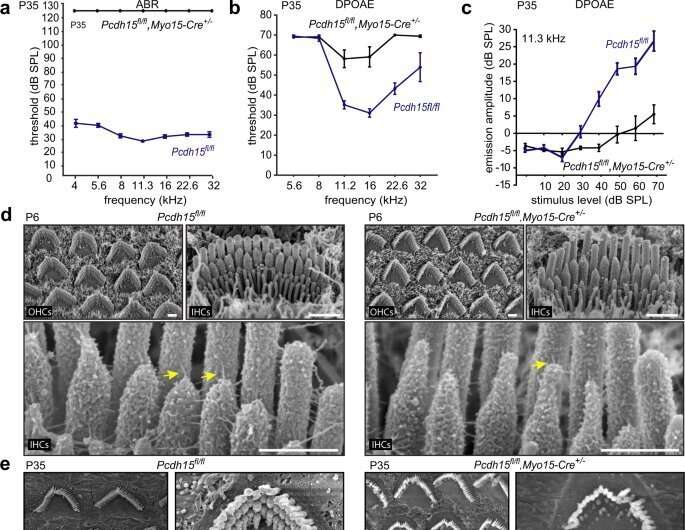
Immagine: topi knockout condizionali Myo15-Cre mostrano una patologia cocleare funzionale e morfologica ritardata. Credito: comunicazioni della natura (2023). DOI: 10.1038/s41467-023-38038-y-
Se il mini-gene funziona nella retina del pesce zebra, i ricercatori passeranno a testare l’approccio nei primati e, infine, negli esseri umani.
Fonte:Nature