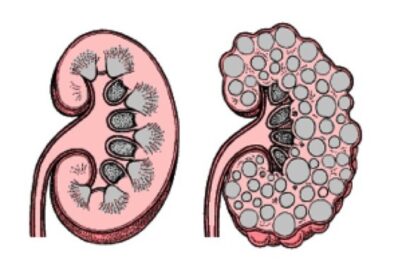
(Malattia del rene policistico-Immagine Credit Publicn Domain).
Si stima che circa 12,5 milioni di persone nel mondo soffrano di malattia del rene policistico autosomico dominante (ADPKD), una delle condizioni monogenetiche più comuni conosciute dall’umanità.
Una caratteristica clinica dell’ADPKD è la crescita incessante di innumerevoli cisti piene di liquido nei reni, che sostituiscono il normale parenchima e, nel corso di decenni, causano un massiccio ingrossamento renale bilaterale e insufficienza renale. L’ADPKD si verifica a causa di mutazioni eterozigoti con perdita di funzione in PKD1 (~ 78% dei casi) o PKD2(~15% dei casi).
L’ipotesi classica per l’inizio della formazione delle cisti è che oltre a una mutazione inattivante della linea germinale in un allele del gene PKD, vi sia un’inattivazione somatica (denominata secondo colpo) nell’altro allele, causando una completa perdita dell’espressione della policistina nella cellula.
Tuttavia, negli ultimi anni, diverse linee di evidenza supportano la soglia di dose del gene come meccanismo coinvolto nella cistogenesi. Questa ipotesi postula che la perdita completa di PKD1 non sia necessaria, ma piuttosto la cistogenesi ne consegue se il dosaggio funzionale di PKD1 scende al di sotto di una soglia critica. Come prova di principio, l’abbassamento della dose di Pkd1 è sufficiente per produrre PKD in topi, maiali e scimmie, secondo il nuovo studio. Pertanto, se il dosaggio ridotto provoca la malattia del rene policistico, aumentando l’espressione della normale allele PKD1 si potrebbe arrestare il disturbo. Tuttavia, nonostante questo potenziale trasformativo, i fattori che regolano il dosaggio di PKD1 nell’ADPKD sono per lo più sconosciuti e attualmente non ci sono meccanismi per attivare il normale allele PKD1.
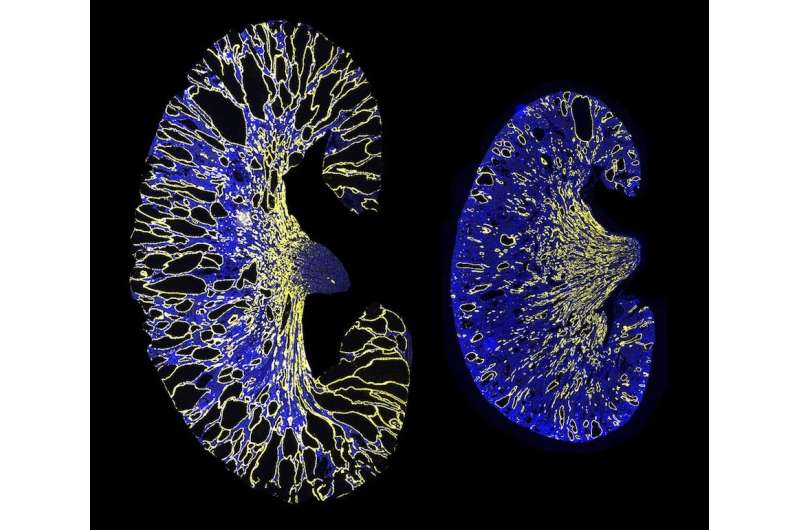
Immagine: un rene gravemente cistico da un modello murino ADPKD (a sinistra). L’eliminazione del sito di legame PKD1 miR-17 riduce notevolmente la crescita della cisti (a destra). Il giallo indica i dotti collettori dei reni e il blu indica i nuclei. Credito: UT Southwestern Medical Center
“Il blocco dell’inibizione dell’espressione genica PKD1 e PKD2 eliminando un sito di legame per i microRNA ha ostacolato la formazione e la crescita di cisti renali nei modelli di malattia del rene policistico autosomico dominante (ADPKD)”, hanno riferito i ricercatori della UT Southwestern.
I risultati dello studio, pubblicati su Nature Communications, suggeriscono una strategia per la terapia genica con il potenziale per arrestare o curare la ADPKD.
“Da più di 25 anni sappiamo che la malattia del rene policistico è causata da mutazioni dei geni PKD1 o PKD2. Tuttavia, non esiste una strategia terapeutica per perseguire queste cause profonde”, ha affermato Vishal Patel, MD, Professore associato di medicina interna nella divisione di Nefrologia presso UTSW e corrispondente autore dell’articolo.
La ADPKD è tra le condizioni genetiche umane più comuni e la causa genetica più frequente di insufficienza renale. Colpisce circa 12,5 milioni di persone in tutto il mondo. E’ una malattia ereditaria in cui i pazienti ereditano tipicamente una copia mutata di PKD1 (o PKD2) e una copia normale.
La malattia è caratterizzata dalla formazione frequente di molte piccole sacche piene di liquido chiamate cisti renali, che si ritiene si formino quando i livelli di PKD1 o PKD2 scendono al di sotto di una soglia critica. Ciò può verificarsi quando la normale copia del gene non produce una quantità sufficiente di proteine Polycystin-1/Polycystin-2.
Vedi anche:Rene policistico: la melatonina efficace contro la malattia
Le proteine sono prodotte (o tradotte) dall’acido ribonucleico messaggero di un gene (mRNA). A un’estremità del filamento di mRNA c’è una regione di codice che aiuta a proteggerlo dalla degradazione, ma può anche controllare la quantità di proteina prodotta. Il legame dei microRNA a questa regione del codice mRNA può bloccare la traduzione, portando alla produzione di meno proteine.
PKD1 contiene un sito di legame per miR-17, un microRNA altamente espresso e attivo nei modelli di ADPKD. Quindi, il Dr. Patel e i suoi colleghi si sono chiesti se il blocco del legame di miR-17 con PKD1 potesse prevenire la formazione di cisti renali.
I ricercatori hanno eliminato il sito di legame del miR-17 dall’mRNA di PKD1 in colture cellulari e un modello murino di ADPKD. I loro risultati hanno indicato che l’eliminazione del sito di legame aumentava la stabilità del filamento di mRNA, aumentava i livelli di policistina-1 e riduceva la crescita delle cisti renali. Inoltre, il gruppo ha scoperto che il blocco del legame di miR-17 all’mRNA di PKD1 con un farmaco anti-miR-17 dopo la formazione di cisti riduceva anche la crescita delle cisti, indicando che questa interazione potrebbe essere un bersaglio promettente per il trattamento della malattia del rene policistico (PKD).
“Ci sono numerose condizioni genetiche in cui una copia del gene causale è mutata, ma l’altra copia è ancora normale. Il nostro approccio per sfruttare la copia normale rimanente è probabilmente applicabile a molte altre malattie oltre alla malattia del rene policistico”, ha affermato il Dr. Patel.
Fonte:Nature