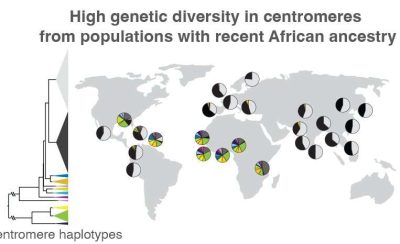
(Genoma-immagine: quando i ricercatori hanno confrontato le regioni centromeriche di 1.600 persone in tutto il mondo, hanno scoperto che quelle senza origini africane recenti avevano principalmente due tipi di variazioni di sequenza. Le proporzioni di queste due variazioni sono rappresentate dai cunei neri e grigio chiaro all’interno dei cerchi, che sono posti sulla mappa vicino al luogo in cui ogni gruppo di individui è stato campionato. Quelli dell’Africa o di altre aree con una grande percentuale di persone con origini africane recenti, come i Caraibi, avevano una variazione della sequenza centromerica molto più ampia, rappresentata dai cunei multicolori. Tali variazioni potrebbero aiutare a tenere traccia di come si evolvono le regioni centromeriche, nonché di come queste varianti genetiche sono correlate alla salute e alla malattia. Credito: Nicolas Altemose, UC Berkeley).
Quando gli scienziati hanno annunciato la sequenza completa del genoma umano nel 2003, stavano un po’ sbagliando.
Infatti, quasi 20 anni dopo, circa l’8% del genoma non è mai stato completamente sequenziato, soprattutto perché è costituito da frammenti di DNA altamente ripetitivi che sono difficili da allineare con il resto. Ma un consorzio di tre anni ha finalmente riempito quel DNA rimanente, fornendo la prima sequenza genomica completa e priva di gap a cui scienziati e medici possono fare riferimento.
Il genoma appena completato, chiamato T2T-CHM13, rappresenta un importante aggiornamento rispetto all’attuale genoma di riferimento, chiamato GRCh38, che viene utilizzato dai medici durante la ricerca di mutazioni legate alla malattia, nonché dagli scienziati che osservano l’evoluzione della variazione genetica umana.
Tra le altre cose, le nuove sequenze di DNA rivelano dettagli mai visti prima sulla regione intorno al centromero, che è dove i cromosomi vengono afferrati e separati quando le cellule si dividono, assicurando che ogni cellula “figlia” erediti il numero corretto di cromosomi. La variabilità all’interno di questa regione può anche fornire nuove prove di come i nostri antenati umani si siano evoluti in Africa.
Vedi anche:Completata la mappa del genoma umano
“Scoprire la sequenza completa di queste regioni del genoma precedentemente mancanti ci ha detto molto su come sono organizzate, cosa totalmente sconosciuta per molti cromosomi”, ha detto Nicolas Altemose, un borsista post-dottorato presso l’Università della California, Berkeley e coautore di quattro nuovi articoli sul genoma completato. “Prima avevamo solo l’immagine più sfocata di ciò che c’era, e ora è cristallina fino alla risoluzione di una singola coppia di basi”.
Altemose è il primo autore di un articolo che descrive le sequenze delle coppie di basi attorno al centromero. Un articolo che spiega come è stato eseguito il sequenziamento apparirà nell’edizione cartacea del 1 aprile della rivista Science , mentre il documento sul centromero di Altemose e altri quattro che descrivono ciò che ci dicono le nuove sequenze sono riassunti nella rivista con i documenti completi pubblicati online. Quattro articoli complementari, incluso uno di cui Altemose è co-primo autore, appariranno anche online il 1° aprile sulla rivista Nature Methods.
Il sequenziamento e l’analisi sono stati eseguiti da un team di oltre 100 persone, il cosiddetto Telemere-to-Telomere Consortium o T2T, dal nome dei telomeri che ricoprono le estremità di tutti i cromosomi. La versione gapless del consorzio di tutti i 22 autosomi e del cromosoma sessuale X è composta da 3.055 miliardi di paia di basi, le unità da cui sono costruiti i cromosomi e i nostri geni e 19.969 geni codificanti proteine. Dei geni codificanti le proteine, il team T2T ne ha trovati circa 2.000 nuovi, la maggior parte dei quali disabilitati, ma 115 dei quali possono ancora essere espressi. Hanno anche trovato circa 2 milioni di varianti aggiuntive nel genoma umano, 622 delle quali si trovano in geni clinicamente rilevanti.
“In futuro, quando qualcuno avrà il proprio genoma sequenziato, saremo in grado di identificare tutte le varianti nel suo DNA e utilizzare tali informazioni per guidare meglio la propria assistenza sanitaria”, ha affermato Adam Phillippy, uno dei leader di T2T e ricercatore seniuor presso il National Human Genome Research Institute (NHGRI) del National Institutes of Health. “Completare davvero la sequenza del genoma umano è stato come indossare un nuovo paio di occhiali. Ora che possiamo vedere tutto chiaramente, siamo un passo più vicini alla comprensione del significato di tutto ciò”.
Il centromero in evoluzione
Le nuove sequenze di DNA dentro e intorno al centromero ammontano a circa il 6,2% dell’intero genoma, o quasi 190 milioni di coppie di basi o nucleotidi. Delle restanti sequenze aggiunte di recente, la maggior parte si trova intorno ai telomeri alla fine di ciascun cromosoma e nelle regioni circostanti i geni ribosomiali. L’intero genoma è composto da soli quattro tipi di nucleotidi che, in gruppi di tre, codificano per gli amminoacidi utilizzati per costruire le proteine. La ricerca principale di Altemose riguarda la ricerca e l’esplorazione di aree dei cromosomi in cui le proteine interagiscono con il DNA.
“Senza proteine, il DNA non è niente“, ha detto Altemose, che ha conseguito un dottorato di ricerca in bioingegneria congiuntamente dalla UC Berkeley e UC San Francisco nel 2021 dopo aver ricevuto un D.Phil in statistica dell’Università di Oxford. “Il DNA è un insieme di istruzioni che nessuno legge se non ha proteine in giro per organizzarlo, regolarlo, ripararlo quando è danneggiato e replicarlo. Le interazioni proteina-DNA sono davvero dove si svolge tutta l’azione per la regolazione del genoma ed essere in grado di mappare dove determinate proteine si legano al genoma è davvero importante per comprendere la loro funzione”.
Dopo che il consorzio T2T ha sequenziato il DNA mancante, Altemose e il suo team hanno utilizzato nuove tecniche per trovare il punto all’interno del centromero in cui un grande complesso proteico chiamato cinetocore afferra saldamente il cromosoma in modo che altre macchine all’interno del nucleo possano separare le coppie di cromosomi.
“Quando questo processo va storto, finisci con i cromosomi non segregati e questo porta a tutti i tipi di problemi“, ha detto il ricercatore. “Se ciò accade nella meiosi, significa che puoi avere anomalie cromosomiche che portano ad aborto spontaneo o malattie congenite. Se accade nelle cellule somatiche, puoi finire con il cancro, fondamentalmente, cellule che hanno una massiccia cattiva regolazione”.
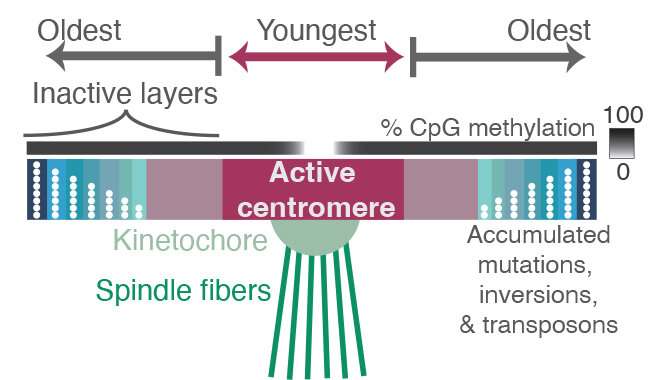
Quello che i ricercatori hanno trovato dentro e intorno ai centromeri erano strati di nuove sequenze che si sovrapponevano a strati di sequenze più vecchie, come se attraverso l’evoluzione nuove regioni del centromero fossero state stabilite ripetutamente per legarsi al cinetocore. Le regioni più vecchie sono caratterizzate da mutazioni e delezioni più casuali, indicando che non sono più utilizzate dalla cellula. Le sequenze più recenti in cui si lega il cinetocore sono molto meno variabili e anche meno metilate. L’aggiunta di un gruppo metilico è un tag epigenetico che tende a silenziare i geni.
Tutti gli strati dentro e intorno al centromero sono composti da lunghezze ripetitive di DNA, basate su un’unità lunga circa 171 paia di basi, che è all’incirca la lunghezza del DNA che avvolge un gruppo di proteine per formare un nucleosoma, mantenendo il DNA impacchettato e compatto. Queste 171 unità di coppie di basi formano strutture ripetute ancora più grandi che vengono duplicate molte volte in tandem, costruendo un’ampia regione di sequenze ripetitive attorno al centromero.
Il team di T2T si è concentrato su un solo genoma umano, ottenuto da un tumore non canceroso chiamato talpa idatiforme, che è essenzialmente un embrione umano che ha rifiutato il DNA materno e ha invece duplicato il suo DNA paterno. Tali embrioni muoiono e si trasformano in tumori. Ma il fatto che questa talpa avesse due copie identiche del DNA paterno – entrambe con il cromosoma X del padre, invece di DNA diverso da madre e padre – ha reso più facile il sequenziamento.
I ricercatori hanno anche rilasciato questa settimana la sequenza completa di un cromosoma Y da una fonte diversa, che ha impiegato quasi tutto il tempo per assemblarsi quanto il resto del genoma messo insieme, ha detto Altemose. L’analisi di questa nuova sequenza del cromosoma Y apparirà in una pubblicazione futura.
Altemose e il suo team, che includeva la scienziata del progetto UC Berkeley Sasha Langley, hanno anche utilizzato il nuovo genoma di riferimento come un’impalcatura per confrontare il DNA centromerico di 1.600 individui provenienti da tutto il mondo, rivelando importanti differenze sia nella sequenza che nel numero di copie del DNA ripetitivo intorno il centromero. Studi precedenti hanno dimostrato che quando gruppi di antichi umani migrarono dall’Africa al resto del mondo, portavano con sé solo un piccolo campione di varianti genetiche. Altemose e il suo team hanno confermato che questo pattern si estende ai centromeri.
“Quello che abbiamo scoperto è che negli individui con ascendenza recente al di fuori del continente africano, i loro centromeri, almeno sul cromosoma X, tendono a cadere in due grandi gruppi, mentre la maggior parte della variazione interessante è negli individui che hanno ascendenza africana recente”, spiega Altemose. “Questa non è del tutto una sorpresa, dato quello che sappiamo sul resto del genoma. Ma quello che suggerisce è che se vogliamo guardare l’interessante variazione in queste regioni centromeriche, dobbiamo davvero fare uno sforzo mirato per sequenziare più genomi africani e completare l’assemblaggio della sequenza da telomero a telomero”.
“Le sequenze di DNA attorno al centromero potrebbero anche essere utilizzate per risalire ai lignaggi umani fino ai nostri antenati scimmie comuni”, ha osservato il ricercatore.”Man mano che ci si allontana dal sito del centromero attivo, si ottiene una sequenza sempre più degradata, al punto che se ci si allontana sulle rive più lontane di questo mare di sequenze ripetitive, si inizia a vedere l’antico centromero che, forse nei nostri lontani antenati primati era solito legarsi al cinetocore”, ha detto Altemose. “È quasi come strati di fossili”.
Sequenza di lettura lunga un punto di svolta
Il successo del T2T è dovuto al miglioramento delle tecniche per il sequenziamento simultaneo di lunghi tratti di DNA, che aiuta a determinare l’ordine di tratti di DNA altamente ripetitivi. Tra questi ci sono il sequenziamento HiFi di PacBio, che può leggere lunghezze di oltre 20.000 coppie di basi con elevata precisione. La tecnologia sviluppata da Oxford Nanopore Technologies Ltd., d’altra parte, può leggere fino a diversi milioni di coppie di basi in sequenza, anche se con una fedeltà inferiore. Per fare un confronto, il cosiddetto sequenziamento di nuova generazione di Illumina Inc. è limitato a centinaia di coppie di basi.
“Queste nuove tecnologie di sequenziamento del DNA a lunga lettura sono semplicemente incredibili; sono rivoluzionarie, non solo per questo mondo ripetitivo del DNA, ma perché consentono di sequenziare singole lunghe molecole di DNA”, ha affermato Altemose. “Puoi iniziare a porre domande a un livello di risoluzione che prima non era possibile, nemmeno con metodi di sequenziamento a lettura breve”.
Altemose intende esplorare ulteriormente le regioni centromeriche, utilizzando una tecnica migliorata che lui e colleghi di Stanford hanno sviluppato per individuare i siti sul cromosoma che sono legati dalle proteine, in modo simile a come il cinetocore si lega al centromero. Anche questa tecnica utilizza la tecnologia di sequenziamento a lettura lunga. Lui e il suo gruppo hanno descritto la tecnica, chiamata Directed Methylation with Long-read sequencing (DiMeLo-seq), in un articolo apparso questa settimana sulla rivista Nature Methods.
Nel frattempo, il consorzio T2T sta collaborando con lo Human PanGenome Reference Consortium per lavorare verso un genoma di riferimento che rappresenti l’intera umanità.
“Invece di avere solo un riferimento da un individuo umano o da una talpa idatiforme, che non è nemmeno un vero individuo umano, dovremmo avere un riferimento che rappresenti tutti”, ha detto Altemose. “Ci sono varie idee su come realizzarlo. Ma ciò di cui abbiamo bisogno prima è una comprensione dell’aspetto di quella variazione e abbiamo bisogno di molte sequenze genomiche individuali di alta qualità per ottenerlo”.
Il suo lavoro sulle regioni centromeriche, che ha definito “un progetto di passione”, è stato finanziato da borse di studio post-dottorato. I leader del progetto T2T erano Karen Miga dell’UC Santa Cruz, Evan Eichler dell’Università di Washington e Adam Phillippy dell’NHGRI, che ha fornito gran parte del finanziamento. Altri coautori dell’articolo sul centromero della UC Berkeley sono Aaron Streets, assistente Professore di bioingegneria; Abby Dernburg e Gary Karpen, Professori di biologia molecolare e cellulare; lo scienziato del progetto Sasha Langley e l’ex borsista post-dottorato Gina Caldas.
Fonte:Science