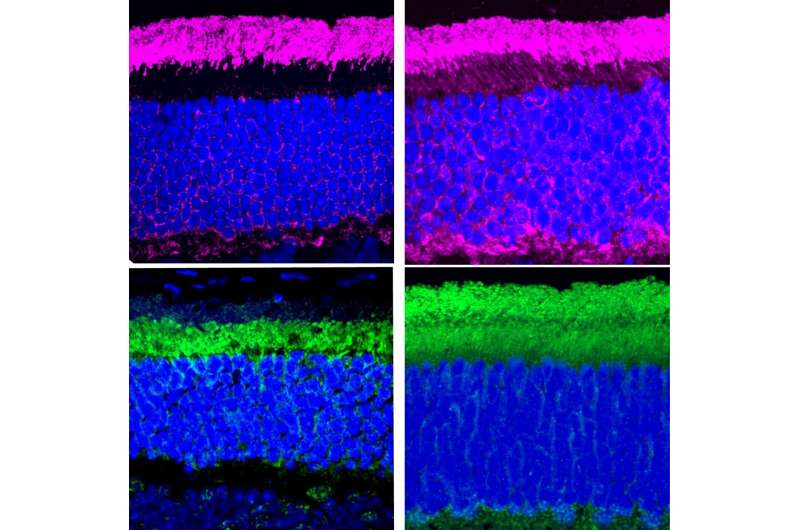
(Sindrome di Uscher-Immagine: nel tessuto oculare di un modello murino con sindrome di Usher (a destra), le proteine del ciclo luce-buio (viola e verde) si trovano sparse nei due compartimenti dei fotorecettori anziché separate nell’uno o nell’altro come nel tessuto oculare sano del topo (a sinistra). Credito: eLife).
La sindrome di Usher di tipo 1F (USH1F) è una rara malattia ereditaria che colpisce udito, vista ed equilibrio. Causa una perdita dell’udito da lieve a grave, che in alcuni casi può essere combinata con un deficit di equilibrio e una perdita della vista che peggiora nel tempo, portando alla cecità. Attualmente non sono disponibili terapie. Tra gli ebrei ashkenaziti, c’è una probabilità del 2% che ogni persona sia portatrice della mutazione di tipo 1F della sindrome di Usher, che rappresenta circa il 60% dei casi di sindrome di Usher di tipo 1. Non esistono terapie approvate per prevenire la perdita della vista o per ripristinare la vista nelle persone con sindrome di Usher.
Utilizzando i dati generati da pazienti e topi con mutazione genetica per la sindrome di Usher, i ricercatori della University of Maryland School of Medicine (UMSOM), del National Eye Institute (NEI) del National Institutes of Health e del National Institute on Deafness and Other Communication Disorders (NIDCD), hanno documentato la storia naturale della disabilità visiva nei pazienti e ha identificato il meccanismo cellulare alla base della progressiva perdita della vista.
Sulla base di questi risultati, pubblicati il 9 novembre 2021, sulla rivista eLife, il team è stato in grado di testare una terapia con retinoidi che ha migliorato la vista nei topi con sindrome di Usher. I ricercatori hanno affermato che la valutazione di una terapia simile dovrebbe ora essere presa in considerazione nelle persone con sindrome di Usher.
“Il farmaco che abbiamo usato nei topi può fornire un primo passo per migliorare la salute degli occhi nelle persone con sindrome di Usher di tipo 1F”, ha affermato Zubair M. Ahmed, Ph.D., Professore di otorinolaringoiatria, chirurgia della testa e del collo e oftalmologia presso l’UMSOM. “Sfortunatamente, questi farmaci non cureranno in modo permanente la perdita della vista, poiché il farmaco non ripara i danni o previene la degenerazione degli occhi. Tuttavia, dovrebbe migliorare la funzione del tessuto che questi pazienti hanno ancora”.
Il primo autore dello studio Saumil Sethna, Ph.D., Instructor in Otorinolaringoiatria—Head & Neck Surgery, ha dichiarato: “Ci sono attualmente parenti approvati dalla FDA di questi farmaci retinoidi che sono disponibili e hanno superato studi clinici per la sicurezza, insieme ad altri che sono in studi clinici di fase II per il trattamento di altri tipi di disturbi della perdita della vista”.
Il team spera di collaborare con una delle aziende che testano questi farmaci per avviare una sperimentazione clinica in pazienti con sindrome di Usher di tipo 1F per verificare se il nuovo farmaco può aiutare a prevenire la continua perdita della vista.
“L’identificazione di una mutazione chiave nel gene PCDH15 quasi due decenni fa è stata una svolta fondamentale, facilitando la diagnosi e lo screening per una certa forma di sindrome di Usher, che ora porta alla scoperta di una potenziale terapia preventiva per la perdita della vista associata alla sindrome“, ha detto Thomas B. Friedman, Ph.D., Capo del Laboratorio di Genetica Molecolare presso il NIDCD. “Questo lavoro esemplifica il valore della ricerca scientifica di base nel guidare lo sviluppo di nuovi strumenti diagnostici e terapeutici”.
Prima di questo studio, erano stati riportati solo dati aneddotici per la sindrome di Usher di tipo 1F senza alcuna analisi dettagliata dei dati del peggioramento delle anomalie oculari nel tempo. All’inizio degli anni 2000, i coautori di questo studio, tra cui il Dott. Friedman del NIDCD, hanno avviato un progetto di ricerca sulla sindrome di Usher e hanno arruolato 13 partecipanti allo studio con sindrome di Usher 1F per seguire la progressione naturale della loro cecità per 20 o più anni. All’epoca il Dottor Ahmed era un borsista post-dottorato nel laboratorio del Dottor Friedman. Wadih Zein, MD, un oftalmologo NEI con esperienza nelle degenerazioni retiniche ereditarie ed è stato il ricercatore principale nello studio e ha riassunto i risultati dei pazienti raccolti nel corso degli anni.
Vedi anche:Una tecnica di iniezione cellulare potrebbe aiutare a invertire la perdita della vista
Separatamente, il Dr. Ahmed ha creato un topo con una sindrome di Usher trovata in 13 pazienti del Dr. Friedman. Questa mutazione nel gene PCDH15 porta a una versione abbreviata della proteina protocaderina-15. Tuttavia, il meccanismo per cui questa protocaderina-15 mutante ha portato alla cecità era sconosciuto. Per convalidare questa ipotesi i team del Dr. Zein del Dr. Friedman e del Dr. Ahmed hanno deciso di collaborare e confrontare le caratteristiche degli occhi di umani e topi con questa condizione genetica che avevano raccolto in modo indipendente nel corso degli anni.
Confrontando il modello murino della sindrome di Usher con topi sani, il team del Dr. Ahmed ha identificato due funzioni della protocaderina-15. In primo luogo, la protocaderina-15 aiuta le proteine del ciclo luce-buio a spostarsi avanti e indietro tra i diversi compartimenti dei fotorecettori che rilevano la luce nell’occhio. In secondo luogo, la protocaderina-15 è necessaria anche per riciclare le molecole essenziali per il funzionamento del tessuto oculare, note come retinoidi. I topi mutanti con sindrome di Usherdi tipo 1F avevano livelli ridotti di retinoidi in un certo tipo di cellula oculare.
Successivamente, i ricercatori hanno somministrato un farmaco retinoide ai topi con sindrome di Usher per vedere se migliorava la loro vista. Le iniezioni di retinoidi nei topi con sindrome di Usher hanno aumentato l’attività elettrica negli occhi dei topi più giovani e adulti, indicando un miglioramento della funzione visiva.
“Oltre alla sostituzione dei retinoidi, possiamo anche pensare di sviluppare terapie più permanenti per trattare o prevenire la cecità nelle persone con sindrome di Usher di tipo 1F che possano correggere o sostituire anche le altre funzioni della protocaderina-15”, ha affermato il Dott. Ahmed.
E. Albert Reece, MD, Ph.D., MBA, Executive Vice President for Medical Affairs, UM Baltimore e John Z. e Akiko K. Bowers Distinguished Professor e Dean, University of Maryland School of Medicine hanno dichiarato: “Persone nate con sordità, come le persone con sindrome di Usher, fanno affidamento sugli altri sensi per comunicare e comprendere il mondo. La successiva perdita della vista può rappresentare sfide significative. Questo lavoro costruisce scoperte fondamentali che un giorno potrebbero migliorare la vita delle persone con malattie genetiche rare come la sindrome di Usher”.
Fonte:eLife