Proteine:Immagine:la funzione di una proteina è determinata dalla sua forma 3D. Credito: DeepMind.
Secondo gli scienziati, il programma di apprendimento profondo di Google per determinare le forme 3D delle proteine sta per trasformare la biologia.
Una rete di intelligenza artificiale (AI) sviluppata da DeepMind, derivata dall’intelligenza artificiale di Google, ha fatto un balzo gigantesco nel risolvere una delle più grandi sfide della biologia: determinare la forma 3D di una proteina dalla sua sequenza di amminoacidi.
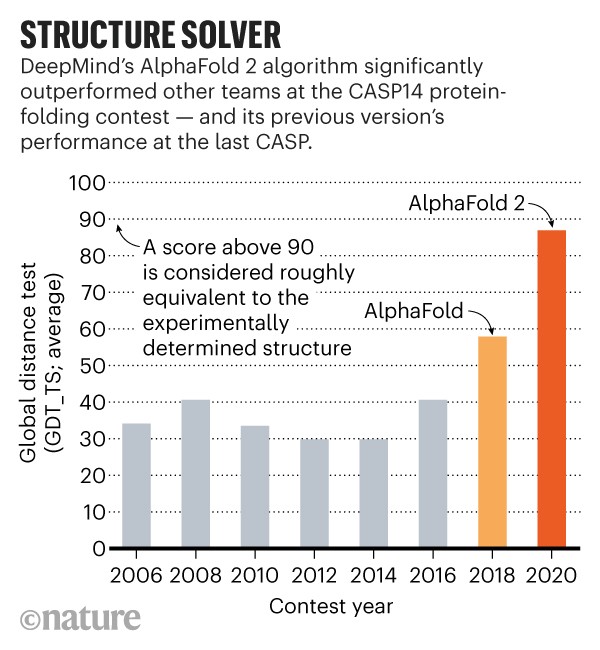
Il programma di DeepMind, chiamato AlphaFold, ha superato circa 100 altri team in una sfida biennale di previsione della struttura proteica chiamata CASP, abbreviazione di Critical Assessment of Structure Prediction.
John Moult, un biologo computazionale presso l’Università del Maryland a College Park, ha co-fondato CASP nel 1994 per migliorare i metodi computazionali per prevedere con precisione le strutture proteiche.
La capacità di prevedere con precisione le strutture proteiche dalla loro sequenza di amminoacidi sarebbe un enorme vantaggio per le scienze della vita e la medicina. Accelererebbe notevolmente gli sforzi per comprendere gli elementi costitutivi delle cellule e consentire una scoperta di farmaci più rapida e avanzata.
AlphaFold è arrivato in cima alla classifica all’ultimo CASP – nel 2018, il primo anno in cui DeepMind con sede a Londra ha partecipato. Ma quest’anno, la rete di apprendimento profondo del gruppo è stata al di sopra delle altre squadre e, dicono gli scienziati, si è comportata così incredibilmente bene da poter annunciare una rivoluzione in biologia.
“È un punto di svolta”, afferma Andrei Lupas, biologo evoluzionista presso il Max Planck Institute for Developmental Biology di Tubinga, in Germania, che ha valutato le prestazioni di diversi team in CASP. AlphaFold lo ha già aiutato a trovare la struttura di una proteina che ha “irritato” il suo laboratorio per un decennio e si aspetta che cambierà il modo in cui lavora e le domande che affronta. “Questa svolta cambierà la medicina. Cambierà la ricerca. Cambierà la bioingegneria. Cambierà tutto “, aggiunge Lupas.
In alcuni casi, le previsioni della struttura di AlphaFold erano indistinguibili da quelle determinate utilizzando metodi sperimentali “gold standard” come la cristallografia a raggi X e, negli ultimi anni, la microscopia crioelettronica (crio-EM). “AlphaFold potrebbe non ovviare alla necessità di questi metodi laboriosi e costosi”, affermano gli scienziati, ma l’IA renderà possibile studiare gli esseri viventi in modi nuovi.
Vedi anche:Il progresso nel campo dell’intelligenza artificiale sarà la chiave per la longevità
Il problema della struttura
Le proteine sono i mattoni della vita, responsabili della maggior parte di ciò che accade all’interno delle cellule. Il modo in cui funziona una proteina e ciò che fa è determinato dalla sua forma 3D: “la struttura è funzione” è un assioma della biologia molecolare. Le proteine tendono ad adottare la loro forma senza aiuto, guidate solo dalle leggi della fisica.
Per decenni, gli esperimenti di laboratorio sono stati il modo principale per ottenere buone strutture proteiche. Le prime strutture complete di proteine furono determinate, a partire dagli anni ’50, utilizzando una tecnica in cui i fasci di raggi X vengono sparati su proteine cristallizzate e la luce diffratta tradotta nelle coordinate atomiche di una proteina. La cristallografia a raggi X ha prodotto la maggior parte delle strutture proteiche. Ma, negli ultimi dieci anni, il cryo-EM, un tipo di microscopia elettronica a trasmissione (TEM) in cui il campione è studiato a temperature criogeniche, è diventato lo strumento preferito di molti laboratori di biologia strutturale.
Gli scienziati si chiedono da tempo come le parti costituenti di una proteina – una serie di diversi amminoacidi – mappino le molte torsioni e pieghe della sua forma finale. “I primi tentativi di utilizzare i computer per prevedere le strutture delle proteine negli anni ’80 e ’90 hanno funzionato male”, affermano i ricercatori. Elevate affermazioni sui metodi negli articoli pubblicati tendevano a disintegrarsi quando altri scienziati li applicavano ad altre proteine.
Moult ha avviato CASP per portare più rigore a questi sforzi. L’evento sfida i team a prevedere le strutture delle proteine che sono state risolte utilizzando metodi sperimentali, ma le strutture non sono state rese pubbliche. Moult attribuisce all’esperimento – non lo chiama una competizione – un enorme miglioramento del campo.
Fonte: DeepMind
“La performance di DeepMind del 2018 al CASP13 ha sorpreso molti scienziati nel campo. Ma il suo approccio era sostanzialmente simile a quello di altri team che stavano applicando l’IA”, afferma Jinbo Xu, biologo computazionale presso l’Università di Chicago, Illinois.
La prima iterazione di AlphaFold ha applicato il metodo AI noto come apprendimento profondo ai dati strutturali e genetici per prevedere la distanza tra coppie di amminoacidi in una proteina. “In una seconda fase che non richiama l’IA, AlphaFold utilizza queste informazioni per elaborare un modello di “consenso” di come dovrebbe apparire la proteina”, afferma John Jumper di DeepMind, che guida il progetto.
“Il team ha cercato di costruire su quell’approccio, ma alla fine non ha avuto i risultati attesi. Quindi ha cambiato rotta”, afferma Jumper, “e ha sviluppato una rete di intelligenza artificiale che incorporava informazioni aggiuntive sui vincoli fisici e geometrici che determinano il modo in cui una proteina si piega”. I ricercatori hanno anche impostato un compito più difficile: invece di prevedere le relazioni tra gli amminoacidi, la rete predice la struttura finale di una sequenza proteica bersaglio. “È un sistema molto più complesso”, dice Jumper.
Precisione sorprendente
CASP si svolge nell’arco di diversi mesi. Le proteine bersaglio o porzioni di proteine chiamate domini – circa 100 in totale – vengono rilasciate regolarmente e i team hanno diverse settimane per presentare le previsioni sulla struttura. Un team di scienziati indipendenti valuta quindi le previsioni utilizzando metriche che misurano quanto sia simile una proteina prevista alla struttura determinata sperimentalmente. I valutatori non sanno chi sta facendo la previsione.
Le previsioni di AlphaFold sono arrivate con il nome di “gruppo 427”, ma la sorprendente accuratezza di molte delle sue voci le ha fatte risaltare, afferma Lupas. “Avevo immaginato che fosse AlphaFold”, dice.
Alcune previsioni erano migliori di altre, ma quasi due terzi erano paragonabili in qualità a strutture sperimentali. “In alcuni casi”, dice Moult, “non era chiaro se la discrepanza tra le previsioni di AlphaFold e il risultato sperimentale fosse un errore di previsione o un artefatto dell’esperimento”.
“Le previsioni di AlphaFold erano scarse corrispondenze con strutture sperimentali determinate da una tecnica chiamata risonanza magnetica nucleare, ma ciò potrebbe dipendere da come i dati grezzi vengono convertiti in un modello”, afferma Moult. “La rete fatica anche a modellare strutture individuali in complessi o gruppi di proteine, per cui le interazioni con altre proteine distorcono le loro forme”.
“Nel complesso, i team hanno previsto le strutture in modo più accurato quest’anno, rispetto all’ultimo CASP, ma gran parte dei progressi può essere attribuita ad AlphaFold”, afferma Moult. “Sugli obiettivi proteici considerati moderatamente difficili, le migliori prestazioni di altre squadre in genere hanno segnato 75 su una scala di 100 punti di accuratezza della previsione, mentre AlphaFold ha ottenuto circa 90 sugli stessi obiettivi”, afferma Moult. “Circa la metà dei team ha menzionato il “deep learning” nell’abstract riassumendo il proprio approccio, suggerendo che l’intelligenza artificiale sta avendo un ampio impatto sul campo. La maggior parte di questi proveniva da team accademici, ma anche Microsoft e l’azienda tecnologica cinese Tencent hanno aderito al CASP14″.
Mohammed AlQuraishi, un biologo computazionale alla Columbia University di New York City e partecipante al CASP, è ansioso di approfondire i dettagli delle prestazioni di AlphaFold al concorso e saperne di più su come funziona il sistema quando il team di DeepMind presenterà il suo approccio il 1° dicembre.
“Penso che sia giusto dire che questo appuntamento sarà dirompente per il campo della previsione della struttura delle proteine. Sospetto che molti lasceranno il campo poiché il problema principale è stato probabilmente risolto “, dice Mohammed AlQuraishi. “È una svolta di prim’ordine, sicuramente uno dei risultati scientifici più significativi della mia vita”.
Strutture più veloci
Una previsione AlphaFold ha contribuito a determinare la struttura di una proteina batterica che il laboratorio di Lupas ha cercato di decifrare per anni. Il team di Lupas aveva precedentemente raccolto dati grezzi di diffrazione dei raggi X, ma la trasformazione di questi modelli simili a Rorschach in una struttura richiede alcune informazioni sulla forma della proteina. I trucchi per ottenere queste informazioni, così come altri strumenti di previsione, erano falliti. “Il modello del gruppo 427 ci ha fornito la nostra struttura in mezz’ora, dopo che noi avevamo trascorso un decennio a provare tutto“, afferma Lupas.
Demis Hassabis, co-fondatore e amministratore delegato di DeepMind, afferma che la società prevede di rendere AlphaFold utile in modo che altri scienziati possano utilizzarlo. (In precedenza ha pubblicato dettagli sufficienti sulla prima versione di AlphaFold per consentire ad altri scienziati di replicare l’approccio). Possono essere necessari giorni ad AlphaFold per elaborare una struttura prevista, che includa stime sull’affidabilità delle diverse regioni della proteina. “Stiamo appena iniziando a capire cosa vorrebbero i biologi”, aggiunge Hassabis, che vede la scoperta di farmaci e la progettazione di proteine come potenziali applicazioni.
All’inizio del 2020, la società ha pubblicato previsioni sulle strutture di una manciata di proteine SARS-CoV-2 che non erano ancora state determinate sperimentalmente. “Le previsioni di DeepMind per una proteina chiamata Orf3a sono finite per essere molto simili a quella determinata successivamente tramite cryo-EM”, afferma Stephen Brohawn, neurobiologo molecolare presso l’Università della California, Berkeley, il cui team ha rilasciato la struttura a giugno. “Quello che sono stati in grado di fare è davvero impressionante”, aggiunge.
Impatto nel mondo reale
È improbabile che AlphaFold ottenga laboratori, come quello di Brohawn, che utilizzano metodi sperimentali per risolvere le strutture proteiche. Ma potrebbe significare che dati sperimentali di qualità inferiore e più facili da raccogliere sarebbero tutto ciò che serve per ottenere una buona struttura. Alcune applicazioni, come l’analisi evolutiva delle proteine, sono destinate a prosperare perché lo tsunami di dati genomici disponibili potrebbe ora essere tradotto in modo affidabile in strutture. “Questo consentirà a una nuova generazione di biologi molecolari di porre domande più avanzate”, afferma Lupas.
La performance di AlphaFold segna anche un punto di svolta per DeepMind. La società è nota soprattutto per il controllo dell’intelligenza artificiale per padroneggiare giochi come Go, ma il suo obiettivo a lungo termine è sviluppare programmi in grado di raggiungere un’intelligenza ampia e simile a quella umana. “Affrontare le grandi sfide scientifiche, come la previsione della struttura delle proteine, è una delle applicazioni più importanti che la sua intelligenza artificiale può realizzare”, afferma Hassabis. “Penso che sia la cosa più significativa che abbiamo fatto, in termini di impatto nel mondo reale”.
Fonte:Nature