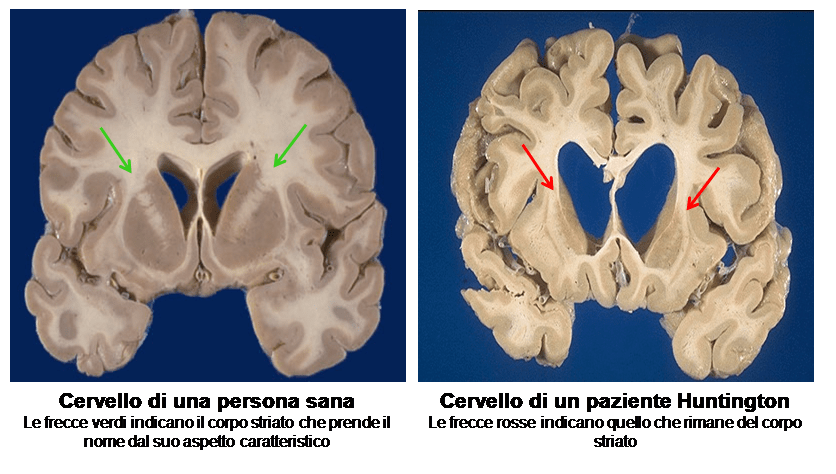
Immagine: Public Domain.
Gli scienziati del Brain Mind Institute dell’EPFL hanno identificato un enzima che può svolgere un ruolo centrale nello sviluppo di una nuova via di trattamento per la malattia di Huntington.
La malattia di Huntington è un disturbo cerebrale progressivo e aggressivamente debilitante che causa movimenti incontrollati, problemi psicologici e perdita della cognizione. È causata da una mutazione nel gene che codifica per la proteina huntingtina, inducendola a costruire una coda anormalmente lunga dell’amminoacido glutammina. Questa coda impedisce all’huntingtina di piegarsi correttamente e di conseguenza si aggrega all’interno dei neuroni del cervello e alla fine li uccide.
L’Huntington colpisce centinaia di migliaia di persone nel mondo e, in quanto malattia “autosomica dominante”, una persona ha bisogno solo di una copia del gene dell’huntingtina mutante per sviluppare la malattia. Scienziati del mondo accademico e dell’industria stanno esplorando approcci diversi per affrontare la malattia. La strategia più popolare è abbassare i livelli di huntingtina o inibirne l’aggregazione o una combinazione di entrambi. Il modo per farlo è “silenziare” il gene dell’huntingtina o attivare meccanismi cellulari che promuovono la degradazione della proteina stessa.
Ora, gli scienziati del laboratorio del Professor Hilal Lashuel all’EPFL hanno identificato un nuovo enzima che fa entrambe le cose. L’enzima, denominato “TBK1“, svolge un ruolo centrale nella regolazione della degradazione e della clearance della proteina huntingtina e introduce modificazioni chimiche che ne bloccano l’aggregazione. “Riteniamo che questo rappresenti un obiettivo praticabile per lo sviluppo di un possibile trattamento della malattia di Huntington”, afferma Lashuel.
L’enzima TBK1 è una “chinasi”. Nella cellula, le chinasi sono enzimi che aggiungono gruppi fosfato a varie biomolecole come proteine o DNA. Nel mondo della cellula, i gruppi fosfato sono portatori di energia, quindi aggiungerne uno essenzialmente “accende” la molecola ricevente.
Studi precedenti hanno dimostrato che l’aggiunta artificiale di gruppi fosfato all’huntingtina può impedirne l’aggregazione e causare la malattia di Huntington. “Tuttavia, per esplorare il potenziale terapeutico della fosforilazione, dovevamo identificare le chinasi naturali che svolgono il lavoro all’interno della cellula”, afferma Lashuel. “Dopo aver esaminato centinaia di chinasi, eravamo entusiasti di identificare TBK1, perché ha svolto il lavoro con elevata specificità ed efficienza”.
I ricercatori hanno scoperto che, quando TBK1 aggiunge un gruppo fosfato ovunque nei primi 17 amminoacidi dell’huntingtina, inibisce la sua capacità di aggregarsi. Questo era il caso sia della versione normale che di quella mutata dell’huntingtina.
Inoltre, l’aumento dei livelli di TBK1 nelle cellule porta a un’eccessiva fosforilazione di uno specifico amminoacido (una serina) nella catena dell’huntingtina. Questo stabilizza la proteina e ne impedisce l’aggregazione.
Infine, è stato anche scoperto che TBK1 segnala alla cellula di degradare e ripulire l’huntingtina prima che si aggreghi. Ciò abbassa i livelli complessivi di huntingtina, che si traduce in una riduzione della formazione di aggregati all’interno della cellula.
Vedi anche:Studio rivela dettagli intricanti sulla proteina della malattia di Huntington
Incoraggiati dalle loro scoperte, gli scienziati sono poi passati a un modello animale della malattia di Huntington: il verme C. elegans. Quello che hanno trovato ha corroborato i loro dati precedenti: sovraesprimere la chinasi TBK1 ha protetto dalla tossicità dell’huntingtina mutante nel verme, prevenendo lo sviluppo della malattia di Huntington. I ricercatori hanno ottenuto risultati simili nei neuroni coltivati.
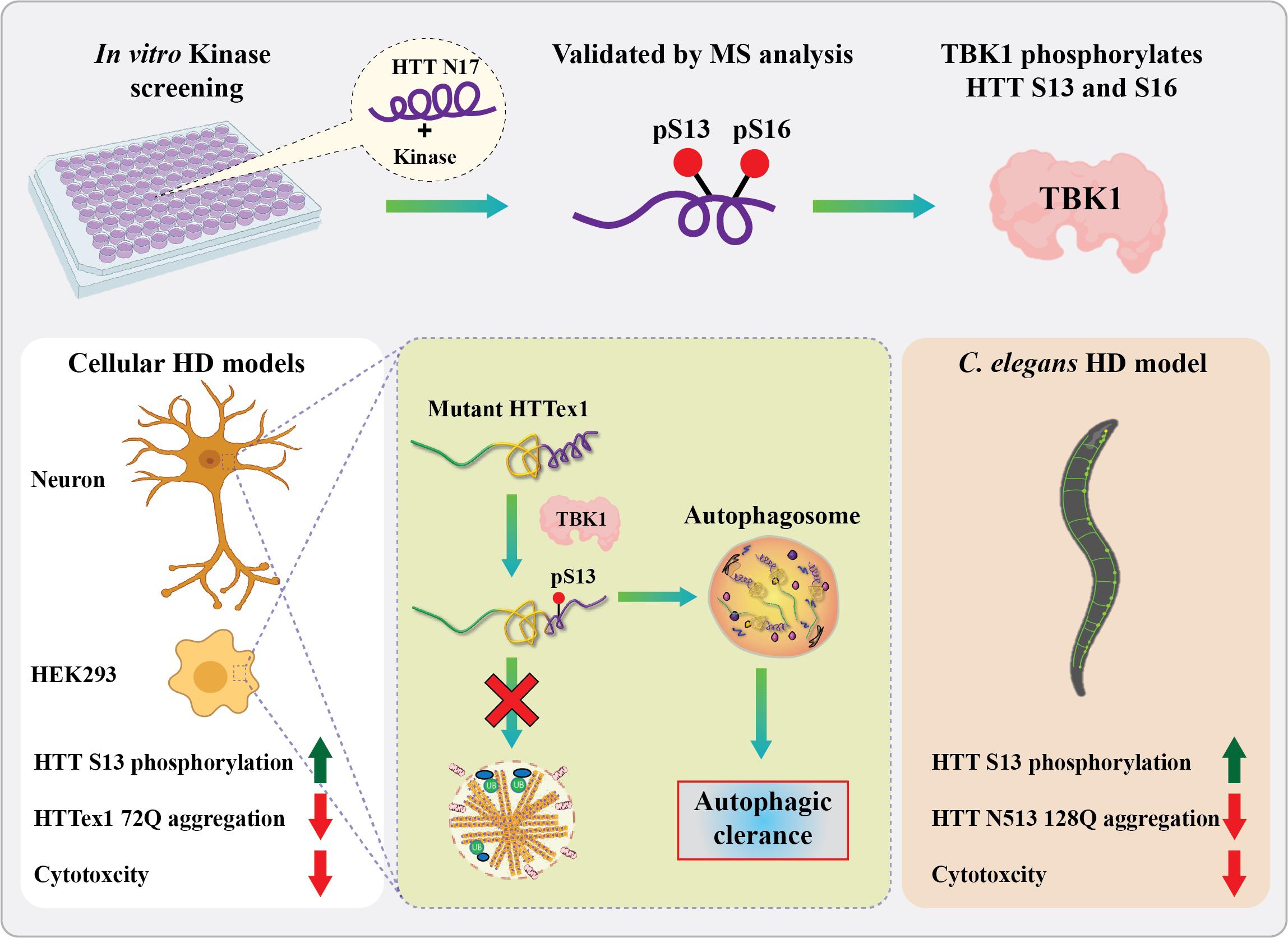
Utilizzando uno screening del cinoma in vitro, il laboratorio di Hilal Lashuel presso l’EPFL ha identificato una nuova chinasi (TBK1) che fosforila la proteina huntingtina a S13 e S16 e ha dimostrato che svolge un ruolo vitale nel regolare la sua aggregazione, clearance e tossicità. Nelle cellule, TBK1 fosforila HTT a S13, porta alla riduzione degli aggregati HTT mutanti nelle cellule e in C. elegans e protegge dalla tossicità HTT tramite un meccanismo che comporta l’aumento della fosforilazione di HTT S13 e / o la promozione della clearance dell’HTT solubile tramite autofagia. Credito: RN Hegde e H. Lashuel (EPFL).
“Il nostro lavoro mostra che l’aumento della fosforilazione mediato da TBK1 e / o la promozione della clearance autofagica dell’huntingtina mutante rappresentano strategie terapeutiche praticabili per il trattamento della malattia di Huntington”, afferma Ramanath Hegde, che ha guidato lo studio.
“Siamo molto entusiasti di questi risultati”, afferma Lashuel. “È stato anche dimostrato che TBK1 regola la clearance e la degradazione delle proteine implicate in altre malattie neurodegenerative. Anche le mutazioni in TBK1 sono state recentemente collegate alla SLA e provocano un’autofagia compromessa, che porta all’accumulo di aggregati. Il nostro obiettivo è trovare piccole molecole o percorsi farmacologici e svilupparli per più malattie neurodegenerative “.
Fonte: Neurologynews