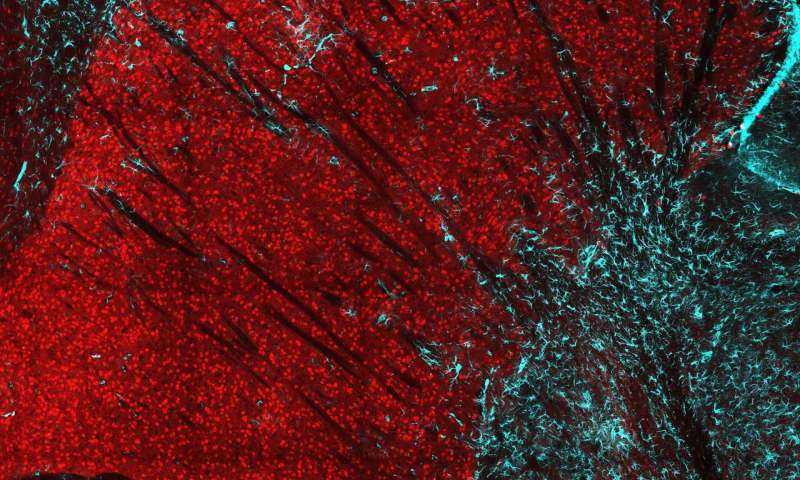
Immagine, una sezione trasversale dello striato nel cervello di un topo. La perdita della proteina huntingtina nei neuroni striatali (rosso) provoca la perdita di neuroni e una risposta infiammatoria, dimostrata dall’infiltrazione di astrociti gliali (ciano). Credito: Caley Burrus, Duke University
Un team di ricerca della Duke University ha identificato una nuova funzione di un gene chiamato huntingtina, il gene che reca la mutazione alla base del disturbo neurodegenerativo progressivo noto come malattia di Huntington.
Utilizzando modelli genetici di topo, i ricercatori hanno scoperto che i neuroni nello striato, un’area cerebrale coinvolta nel controllo dei movimenti, richiedono il gene huntingtina per regolare i movimenti del corpo, mantenere la salute delle cellule durante l’invecchiamento e sviluppare connessioni funzionali tra le cellule.
“Affrontare il ruolo del gene nel mantenere quelle connessioni neurali può fornire una nuova strada contro la malattia di Huntington”, hanno detto i ricercatori.
La malattia di Huntington è una malattia neurodegenerativa ereditaria che di solito emerge nella mezza età e porta a un controllo motorio compromesso, demenza e sintomi psichiatrici. Mentre le basi genetiche di questa malattia letale sono state identificate più di due decenni fa, non ci sono ancora trattamenti approvati per rallentarne la progressione o curarla.
La malattia è causata da una mutazione in una delle due copie del gene huntingtina di una persona. La mutazione provoca la produzione di una versione aberrante della proteina huntingtina, che è tossica per i neuroni. Sebbene la proteina mutante sia espressa in tutto il corpo, i neuroni dello striato sono specificamente vulnerabili ai suoi effetti e degenerano mentre la malattia progredisce.
Mentre la proteina huntingtina mutante è dannosa per i neuroni, può anche interferire con la capacità rimanente, non mutata, di huntingtina di svolgere le sue normali funzioni.
I farmaci attualmente in fase di sperimentazione in studi clinici sono progettati per bloccare la proteina huntingtina difettosa, ma finiscono per ridurre anche la quantità di huntingtina normale nei neuroni. È noto che la huntingtina svolge diverse importanti funzioni nelle cellule, ma il suo ruolo specifico nella salute e nella funzione dei neuroni striatali non era noto.
“Abbiamo ipotizzato che il normale gene della huntingtina svolga un ruolo critico nella salute e nella connettività neuronale e volevamo determinare cosa succede ai neuroni striatali che hanno eliminato la huntingtina“, ha detto l’autore principale dello studio Cagla Eroglu, Professore associato di biologia cellulare e neurobiologia e il co-Direttore della Regeneration Next Initiative di Duke.
Vedi anche, Identificato un meccanismo molecolare coinvolto nella malattia di Huntington
In uno studio pubblicato martedì su Cell Reports, il team ha scoperto che l’eliminazione del gene huntingtina specificamente dai neuroni striatali di topi molto giovani ha causato la morte di questi neuroni con l’invecchiamento dei topi, simile al modello di morte dei neuroni osservati nella malattia di Huntington. I ricercatori hanno anche scoperto che i topi privi di huntingtina nei loro neuroni striatali erano compromessi nella loro capacità di controllare i movimenti. È importante sottolineare che questa perdita di regolazione del movimento è avvenuta anche prima che i neuroni stessi iniziassero a morire.
“Questi risultati suggeriscono che la morte cellulare stessa potrebbe non essere l’unico fattore scatenante dei sintomi della malattia di Huntington”, ha detto Eroglu.
In un cervello sano, i neuroni striatali controllano il movimento comunicando con altri neuroni attraverso connessioni chiamate sinapsi. I ricercatori hanno scoperto che i neuroni striatali privi di huntingtina formavano connessioni sinaptiche anomale, che potrebbero potenzialmente spiegare la problematica funzione motoria dei topi.
“Riteniamo che i cambiamenti a livello neuronale e sinaptico che si verificano prima della morte cellulare contribuiscano alla progressione della malattia“, ha affermato Caley Burrus, Ph.D. ricercatore nel laboratorio di Eroglu e primo autore dello studio.
“È possibile“, hanno affermato gli autori, “che le terapie per affrontare i difetti a livello di sinapsi possano essere utili per i pazienti Huntington”. Il team sta continuando la ricerca studiando il ruolo preciso che la huntingtina svolge nello sviluppo delle sinapsi, studio che potrebbe portare a farmaci specifici progettati per affrontare questi deficit.
“Comprendere i ruoli critici che la huntingtina svolge nei neuroni è essenziale per progettare terapie che aiuteranno le persone con la malattia di Huntington nel modo più sicuro ed efficace possibile”, ha detto Eroglu.
Fonte: Cell Reports