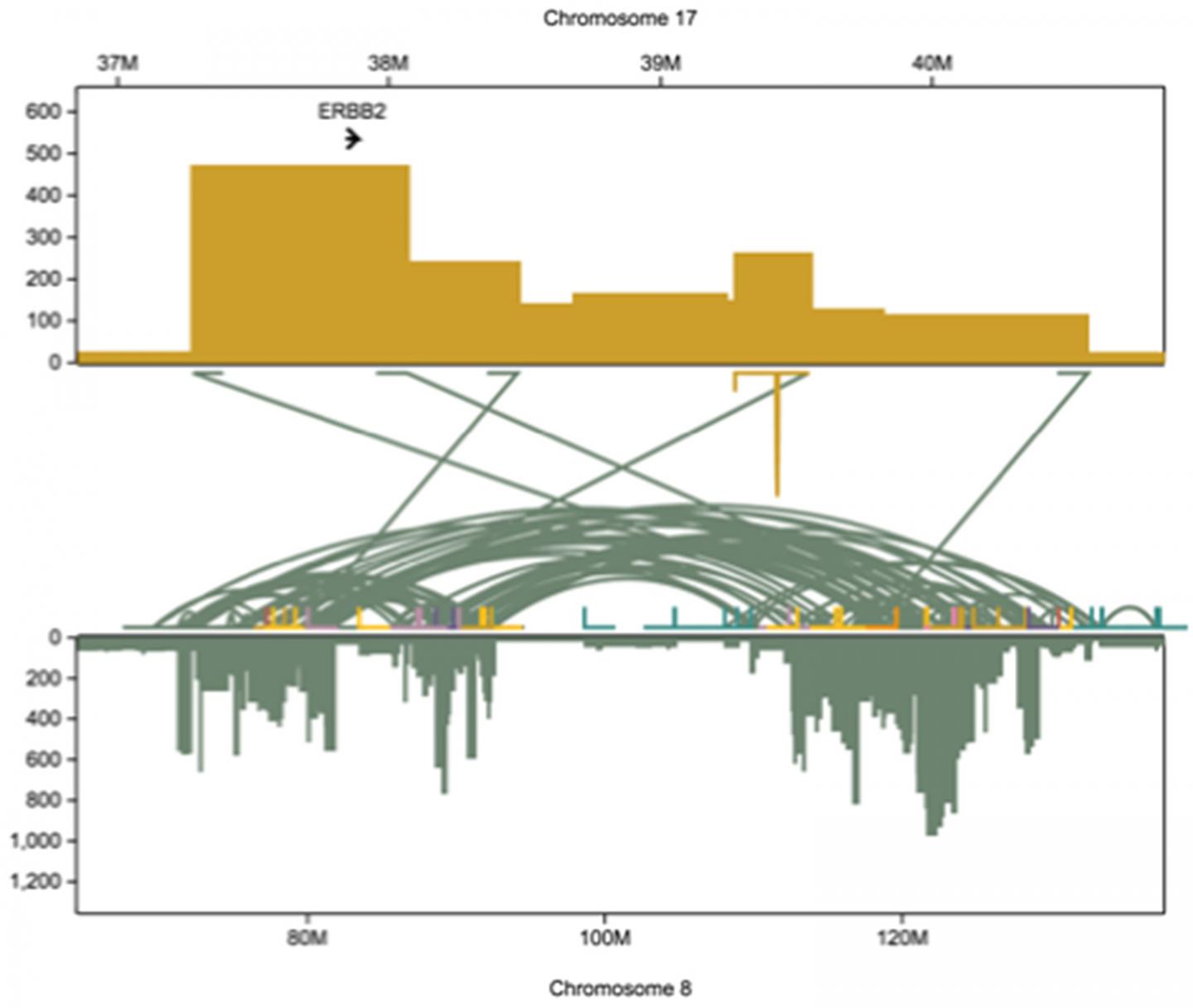
Immagine: “il sequenziamento a lunga lettura ha consentito al team di ricostruire in modo dettagliato la storia di come il gene HER2 viene amplificato in maniera massiccia nelle cellule di carcinoma mammario HER2-positive, afferma il dott. Schatz. Il rettangolo in alto mostra un segmento di 2 milioni di paia di basi del cromosoma 17 occupato dal gene HER2 (chiamato anche ERBB2). Un piccolo segmento del gene, già amplificato in modo massivo, si spezza e si fonde con il cromosoma 8 (rettangolo inferiore). Su quel cromosoma, parti del gene vengono copiate fino a 1000 volte, con vari segmenti che saltano all’interno del cromosoma (archi verdi). Questo dimostra perché vogliamo identificare i pazienti HER2-positivi il prima possibile, per prevenire il tipo di caos che registriamo qui cumulativamente”, dice Schatz. Credit: Schatz Lab, CSHL / JHU
I ricercatori hanno svelato il devastante genoma del cancro al seno.
Nelle cellule tumorali, gli errori genetici causano il caos così come le variazioni strutturali – riarrangiamenti di DNA su vasta scala che possono comprendere grossi blocchi di cromosomi – che disturbano meccanismi attentamente bilanciati che si sono evoluti per regolare la crescita cellulare. I geni che normalmente sono silenti vengono massivamente attivati e si formano le proteine mutanti. Queste e altre interruzioni causano una miriade di problemi che fanno crescere le cellule in modo incontrollato, il segno caratteristico del cancro.
Questa settimana, gli scienziati del Cold Spring Harbor Laboratory (CSHL) hanno pubblicato su Genome Research, una delle mappe più dettagliate mai fatte di variazioni strutturali nel genoma di una cellula cancerosa. La mappa rivela circa 20.000 variazioni strutturali, alcune delle quali non erano mai state notate a causa dei limiti tecnologici.
Il team, guidato dagli esperti di sequenziamento Michael C. Schatz e W. Richard McCombie, ha letto i genomi delle cellule tumorali con la cosiddetta tecnologia di sequenziamento a lungo termine. Questa tecnologia legge segmenti molto più lunghi del DNA rispetto alla vecchia tecnologia a breve lettura. Nei risultati interpretati con due sofisticati pacchetti software pubblicati di recente dal team, sono evidenti due vantaggi: il sequenziamento a lunga lettura è più ricco in termini di informazioni e contesto. Può, ad esempio, avere un senso migliore di tratti ripetitivi di lettere di DNA – che pervadono il genoma – in parte osservandoli all’interno di un contesto fisicamente più ampio.
( Vedi anche: Migliaia di pazienti con cancro al seno potrebbero evitare la chemio).
Il team ha dimostrato il potere della tecnologia a lunga lettura usandola per leggere i genomi delle cellule derivate da una linea cellulare chiamata SK-BR-3, un modello importante per le cellule del cancro al seno con variazioni in un gene chiamato HER2 (a volte chiamato anche ERBB2 ). Circa il 20% dei tumori della mammella sono “HER2-positivi”, ovvero producono una sovrapproduzione della proteina HER2. Questi cancri tendono ad essere tra i più aggressivi.
“La maggior parte delle 20.000 varianti identificate in questa linea cellulare sono state ignorate dal sequenziamento a breve lettura“, afferma Maria Nattestad, che ha svolto il lavoro con i colleghi mentre era ancora membro del laboratorio Schatz presso la CSHL e la Johns Hopkins University. “Di particolare interesse, abbiamo trovato un insieme molto complesso di variazioni del DNA che circondano il gene HER2“.
Nella sua analisi, il team ha combinato i risultati del sequenziamento a lunga lettura con i risultati di un altro tipo di esperimento che legge i messaggi, o trascrizioni, che vengono generati dai geni attivati. Questo quadro più completo ha fornito un resoconto straordinariamente dettagliato di come le variazioni strutturali distruggono il genoma nelle cellule tumorali e fa luce su come le cellule tumorali si evolvono rapidamente.
Schatz, Adjunct Associate Professor presso CSHL e Bloomberg Distinguished Associate Professor presso la Johns Hopkins University, e McCombie, un Professore di CSHL, affermano che è essenziale continuare a costruire un catalogo di varianti di tipi di cellule cancerogene utilizzando le migliori tecnologie disponibili. “Quello utilizzato in questa ricerca è uno strumento inestimabile per catturare la complessità delle variazioni strutturali, quindi ci aspettiamo un’adozione diffusa per l’uso nella ricerca e nella pratica clinica, soprattutto perché i costi di sequenziamento diminuiscono ulteriormente “.
Fonte: EurekAlert